固相化 pH 梯度双向凝胶电泳实验
最新修订时间:
简介
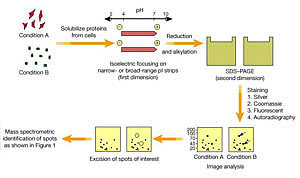
双向电泳是研究蛋白质组学的一种有效方法。与单向电泳相比,它能从复杂的蛋白质混合物中分离出更多的成分。电泳时,蛋白质迁移速度取决于它们的分子形状、大小及所带电荷多少,双向电泳利用后两项特征使蛋白质能够有效分离。
[澳] 理查德 J. 辛普森 (Richard J.Simpson) 主编 何大澄 主译
来源:丁香实验
操作方法
1.取一些肝放入研钵中,如果事先没有被冰冻,可用液氮充满研钵来保持肝冰冻。用研杵将肝破碎成小块。 2.称量冰冻肝的小块 10~40 mg,将其转移至在冰上放置的离心小管中。操作应迅速以尽可能减少肝样品的解冻。将剩余的肝储存在一 80°C。 3.在有肝样品的离心管中,每 40 mg 肝加入 1ml 冷的抽提溶液。每 1ml 抽提溶液加 100 mmol/L PMSF。
方案2 双向凝胶电泳真核生物细胞裂解物的制备实验1.从培养皿中转移细胞。. 如果是贴壁细胞,用细胞刮刀从培养皿中刮下细胞,用 5 ml 吸管将细胞和培养基转移到 15 ml 离心管中。 如果是悬浮细胞,直接将细胞和培养基转移到离心管中。 2.480 g、4°C,离心沉淀细胞 5 min。 3.弃去上清,勿搅动沉淀。 要点:操作以下步骤时,所有细胞需保持冰冻状态;不离心或振荡时,保持细胞在
方案3 双向凝胶电泳大肠杆菌裂解液的制备实验1.将培养基装人质量已知的瓶子或管子中,12000 g、4°C 离心 5 min,以沉淀大肠杆菌。 2.弃去上层清液,完全吸干离心管。称量瓶子或管子的毛重,去除皮重以确定大肠杆菌的重量。 3.在冷抽提液中重悬沉淀,每 1g 细胞用 1Oml 抽提液。将细胞悬液转移到塑料圆锥形离心管中。每 1Oml 抽提液加 1 ul (500U)Benzonase 和 100ul 100
方案4 双向凝胶电泳脑脊液蛋白样品的制备实验1.在室温下解冻脑脊液样品。当样品解冻后,旋紧试管盖,再用旋涡混合器振荡试管。 2.样品溶解时,在 15 ml 离心管及盖上标记所处理样品的名称 3.将 1 ml 脑脊液和 9 ml 乙醇加人 15 ml 离心管中混合。 注意:脑脊液可能是生物危险品。将用过的吸头、试管和手套放在适当的容器中,用 1mol/L NaOH 认真地清洗污染表面。 4
方案5 第一向:蛋白质的等电聚焦电泳实验一、IPG 胶条的水化 1.在新制备的或温和解冻的水化液中加 DTT 至终浓度为 2 g/L。加两性电解质载体或 IPG 缓冲液至终浓度为 0.5%~2.0%。其 pH 范围与用于分析的 IPG 胶条的 PH 范围相匹配。 2.若在水化期间将蛋白质样品加到凝胶上,则将样品用水化液稀释(或溶解)至每胶条含 Img 总蛋白。 上样量取决于实验的目的。高丰度蛋白质上样量
方案6 垂直 SDS 平板凝胶的制备:均一凝胶的灌制实验1.安装玻璃板之前,用乙醇浸过的无棉纸巾擦拭玻璃板,以确保无灰尘颗粒粘在玻璃板表面。 实验提示:处理玻璃板的过程中要戴手套,以减少角蛋白和其他蛋白的污染。 2.对齐两块玻璃板,在两边放入 1.0mm 或 1.5 mm 隔片。 3.用恰好足够的压力将两块玻璃板及隔片夹住,使其保持在适当的位置。将其底部立于水平面上,保证两块玻璃板底边及隔片相互齐平,锁紧夹
方案7 垂直SDS 平板凝胶制备:同时灌制多梯度凝胶实验1.安装玻璃板之前,用乙醇浸过的无棉纸巾擦拭玻璃板,确保无灰尘颗粒粘在玻璃板表面。 实验提不:处理玻璃板的过程中要戴手套,以减少角蛋白和其他蛋白的污染。 2.对齐两块玻璃板,在两边放入 1.0mm 或 1.5 mm 隔板。 3.用不脱色的记号笔标记玻璃板距上沿 0.5 cm 处,在一些系统例如 Bio-Rad ProteanII,玻璃板不等长,应确保标
方案9 第二向:蛋白质的 SDS-PAGE 实验1.在 100°C 加热琼脂糖封闭溶液使之熔化。每块凝胶约需 1ml 溶液。完全熔化琼脂糖要花 1Omin,最好在即将平衡 IPG 胶条时进行(方案 8)。琼脂糖熔化后,将其冷却至 40~50°C。 2.在第二向凝胶表面的平板之间用钳子定位平衡后的 IPG 凝胶条,保证塑料支持条靠着其中一块玻璃平板。在平板之间用一把薄的软塑料尺子推动 IPG 胶条,直到 IPG 底边和凝胶表面紧
方案10 胶体考马斯亮蓝染色实验1.将凝胶置于玻璃或塑料培养皿中固定,轻轻摇动至少 1h。 可将凝胶固定过夜。如果凝胶有标记,多个凝胶可在同一培养皿中固定,固定液的体积应足够大,以使所有凝胶都能自由移动。 2.移走固定液,在摇床上用水清洗凝胶 10 min。 3.弃去水,重复洗 2 次。 4.在洗第三次时,将 4 份考马斯亮蓝储备液和 1 份甲醇混合,以制备胶体考马斯亮蓝染液。
方案11 银氨染色1.将凝胶(见方案 9) 放入有水的平皿中。在往复式或定轨式摇床上摇 5 min,同时制备新鲜的固定液 1。 多块凝胶的固定和染色应在分离的平皿中进行。 2.将水从平皿中排尽,在培养皿中用方形塑料纸或聚碳酸酯薄片支持凝胶。勿用戴手套的手和凝胶直接接触。在平皿中倒人足够的固定液 1 以覆盖凝胶,在摇床上摇动 30~60 min。 3.制备新鲜的固定液 2, 将
方案12 与质谱兼容的银染法实验1.将凝胶放在含 250 ml 固定液的平皿中,在摇床上摇动 5 min。如果有多块凝胶要固定,应在单独的平皿中染色。 2.从平皿中排尽固定液,用方形塑料纸或聚碳酸酯薄片支持凝胶。不允许戴手套的手直接和凝胶接触。再加 250 ml 固定液,继续摇 15 min。 3.弃去固定液,在平皿中再次加人 250 ml 敏化液,在往复式或定轨式摇床上摇动 30 min。
方案13 用 SYPRO Ruby 进行荧光染色1.将胶放入聚丙烯塑料平皿中,其中应有足够的固定溶液,使凝胶在平皿中能自由漂浮。在摇床上摇动 30 min。 如果是多块凝胶同时被固定,应在不同的平皿中染色。 2.从平皿中倒出固定液,在平皿中用方形塑料纸或聚碳酸酯薄片支持凝胶。不要用带手套的手接触凝胶。将凝胶放在 SYPRORuby 溶液中染色 90 min,放置^往复式或定轨式摇床,黑暗中温育过夜。 标准大小的
方案14 用 GelCode 磷蛋白染色试剂盒对磷蛋白进行染色实验1.将凝胶放入盛有 50 ml 水的平皿中,在往复式或定轨式摇床上摇动凝胶 1Omin。 2.将凝胶转移到盛有 25 ml 磺基水杨酸溶液的平皿中,摇动 15 mm。 3.再将凝胶放在 25 ml 含氯化钙的磺基水杨酸溶液中,在摇床上摇动 30 min。 4.迅速用清水洗涤凝胶表面以去除连接在其上的氯化钙。 5.将凝胶转移到 25 ml 0.5mol/L
方案15 双向凝胶的槽式转移实验1.测量 SDS-PAGE 凝胶的尺寸,将转移膜剪成与凝胶相适应的尺寸。 槽式转移系统的膜根据需要可以剪成比凝胶大或者小。 2.用水将薄膜润湿或用甲醇将 PVDF 膜润湿。操作要小心,以避免膜不平整及膜下面产生气泡。 3.将湿润的薄膜转移到一个盛有转移缓冲液的平皿中,使膜在其中平衡 5 min。 4.将印迹纸剪切成合适大小,按照转移设备的指导手册进行操作。
方案16 双向凝胶的半干印迹实验1.测量 SDS-PAGE 凝胶的尺寸,将转移膜与 6 层印迹纸剪切成与凝胶同样大小。 2.用水湿润膜或用甲醇湿润 PVDF 膜,缓慢操作,以避免膜不平整或在膜下产生气泡。 3.将湿润的膜转移到盛有转移缓冲液的平皿中,让其在缓冲液中平衡 5 min。 4.将印迹纸、膜与凝胶排列成「三明治」状,要避免产生气泡,因为气泡会阻止蛋白质的转移。最好按照以下方法操作: (1)印迹纸和 B 吴要用转移
方案17 用丽春红 S 染色膜上的蛋白质1.用丽春红 S 染液浸没转移后的膜,轻摇 5 min。 2.弃去染液,用水清洗膜,重复几次,直到蛋白质条带变得清晰。 不要重复使用染料,因为这可能使结果重复性变差。第一次用完后,将染料尽量排尽。 3.用一只软铅笔标记主要蛋白质条带和标准分子量蛋白质条带。 4.继续用水清洗膜,轻摇直到将丽春红 S 染液完全除去。 5.对膜上的蛋白质条带进行分析或保存。
方案18 用考马斯亮蓝 R250 染色膜上的蛋白质1.将转移后的膜浸在考马斯亮蓝染液中,轻摇 5min 。 2.弃去染液,在摇床上用含乙酸的甲醇进行脱色。不要重复使用染液,因为这样会使结果的重复性变差。 3. 用水洗膜,轻摇 5min。
方案19 用印度墨水染色膜上的蛋白质1.将膜浸人吐温-20 溶液中,轻摇 10 min。 2.弃去吐温-20 溶液。 3.重复步骤1与2三次。 4.将膜转人印度墨水中,轻摇染色 2~18 h。 5.弃去染液,用吐温-20 溶液洗膜,轻摇 5 min。
方案20 用胶体金染色膜上的蛋白质1.将膜浸入吐温-20 溶液中,37°C,轻摇 45 min。 2.室温下,用吐温-20 溶液洗膜,轻摇 5 min。 3.弃去洗液,多次重复步骤①和②。 4.室温下在胶体金染液中染膜 2 h,轻摇。 5.用水洗膜,轻摇 5 min。
相关产品推荐




关于丁香通
公司信息
个人用户
企业机构


