



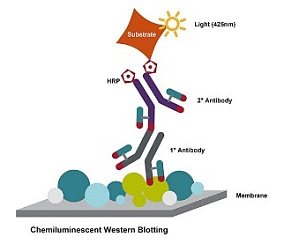
某个实验间隙…萌新:开题报告已提交,转眼 1 个月过去了,实验还是一头雾水,为啥我的 co-IP 拉不下来蛋白?奋斗中的师兄:我当初正好相反,拉下来的蛋白被抗体条带覆盖住了,换个抗体忒不好找,无奈换了个蛋白,还好挺顺利。大师兄:蛋白吧,不但种类多,理化性质差异较大,关键它们又喜欢组队在细胞里干活,IP、co-IP 又是常用的手段,掌握好这个工具还是很重要的,总不能随随便便就换个蛋白。那要怎么快速掌握这项实验,轻松搞定各种疑难杂症呢?我们依据三十多年的 IP/co-IP 实验经验,总结了一线技术支持常被问到的问题,希望这篇纯干货能帮助大家顺利升级打怪,早日驾驭 IP、co-IP。IP、co-IP、pull-down 简介IP(Immunoprecipitation),免疫沉淀:是利用固定在磁珠或琼脂糖树脂等基质上的特异性抗体对抗原进行小型亲和纯化的一种方法Co-IP(co-Immunoprecipitation),免疫共沉淀: co-IP 是一种常用的鉴定蛋白-蛋白相互作用的方法,其基本实验流程和IP类似,通过使用诱饵蛋白(IP 中的抗原,bait protein)特异性抗体间接捕获与其

RNA 干扰(RNAi)是一种转录后基因沉默的机制。同时也是一项成熟的实验技术,它的出现彻底改变了研究人员研究哺乳动物基因表达的方式,并持续为如今的基因功能研究提供有价值的实验结果。RNAi 技术大大提高了在哺乳动物细胞和动物模型中进行基因功能缺失分析的便利性、速度和特异性,因此长期以来一直是深受研究人员信赖的一种选择。小干扰 RNA(siRNA)是 RNAi 技术中最为常用的一种手段,其在基础研究中已经发挥了巨大的作用。回到最基础的实验中,尽管已经非常成熟,想要顺利开展 siRNA 实验并拿到可靠的实验结果却不是那么容易的,除了选择高质量的稳定的siRNA 产品之外,还有很多实验设计和操作方面的注意事项。今天我们为大家总结了成功开展 siRNA 实验的十大秘诀。1.每个基因设计并测试两到四条 siRNA 序列为了找到一个潜在的靶点,分析感兴趣基因的全长寻找氨基酸序列。记录氨基酸和3′ 19核苷酸作为潜在的 siRNA 靶点。潜在的靶点随后通过对 GenBank 数据库的 BLAST 分析进行评估,去掉任何与其他基因有显著同源性的靶序列。如果可能的话,应该针对含有更少二级结构的靶 mR
生成全长转录组使用 Ambion 转录试剂盒合成的大多数 DNA 模板都可以生成全长转录组,无需任何优化。然而,一些模板可能产生过早终止的产物,如较小的离散带或拖尾/降解产物。对于印迹杂交,通常不需要全长 RNA 探针。然而,对于许多其他分析,至关重要的是转录进行到模板的末端,全部生成同一种大下的转录组(例如 NPA,体外翻译研究和结构分析)。转录反应失败或不良的两个最常见因素是标记核苷酸中的抑制剂和质量较差的 DNA 模板。应执行一系列简单的实验来确定转录反应失败的原因;随附的流程图概述了这一过程。以下列出了其他一些策略,用于增加有问题的转录反应中的全长产物比例。增加限制性核苷酸的浓度用最低浓度的标记核苷酸进行转录反应,可能由于核苷酸浓度不足而产生过早终止的转录组。增加限制性核苷酸的浓度通常会提高全长转录组的产量。降低反应的孵育温度通常,转录反应在室温或 37°C 下进行。将温度降低至约 16°C 或甚至 4°C 有时可以改善转录反应。人们认为,较低的反应温度会减慢聚合酶的进展,从而防止其被二级结构或一个特定核苷酸串置换。(想象一下玩具火车全速绕弯前行与缓慢绕同一条路线前行。)使用不
PCR 酶数量如此之多,选择正确的酶可能是一项挑战。用于扩增 DNA 的各种酶的保真性、扩增速度和特异性各不相同。以下三个问题可以帮助您解答选择 PCR 酶时需要重点关注的因素。Q 1:您需要确保序列准确性吗?有时您只需要检测 PCR 产物或估算其大小,例如在对小鼠进行基因分型或筛选重组克隆时。对于这种类型的常规 PCR 分析,您应该使用标准的热稳定 DNA 聚合酶(如 Taq DNA 聚合酶)来确认目的 DNA 存在与否。但是,如果要执行克隆实验或下一代测序(NGS),那么准确性至关重要。为了确保准确进行 DNA 拷贝,请确保选择高保真 DNA 聚合酶。高保真 PCR 酶具有 3'-5’ 核酸外切酶校对活性。当结合错误匹配的碱基对时,DNA 聚合酶停顿,导致合成暂延。合成暂延 允许去除错误匹配的核苷酸并使用正确的核苷酸取代。保持序列准确的 DNA 聚合酶的绝佳选择是 Invitrogen SuperFi 或 Thermo Scientific Phusion 高保真DNA聚合酶。这两种酶具有高保真度,准确度是 Taq DNA 聚合酶的 300 倍或 52 倍,并且具有很高的产率。Q
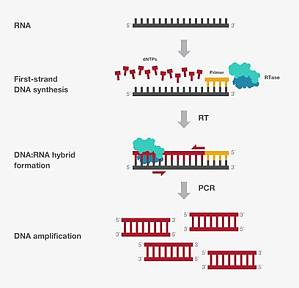
PCR 是众所周知的分子生物学技术之一。20 世纪 70 年代,研究人员首次报道了使用合成引物和 DNA 聚合酶从模板复制单链 DNA。然而直到 1983 年,Kary Mullis 才发明出用于扩增目标 DNA 的研究工具,就是我们今天所熟知的 PCR 方法。从此以后,PCR 成为分子生物学研究必不可少的一部分,被广泛应用于基础研究、疾病诊断、农业检测和法医调查等领域。1.基因表达通常可通过 PCR 来检测不同细胞类型、组织和生物体在特定时间点的基因表达差异。首先,从目标样品中分离出 RNA 并将mRNA逆转录成 cDNA。随后,通过由PCR扩增的cDNA数量,确定 mRNA 的初始水平。这一过程也被称为逆转录 PCR, RT-PCR (图 1)。图 1. RT-PCR.RNA 被逆转录成 cDNA,随后通过 PCR 扩增 cDNA终点 PCR 可通过凝胶里的扩增产物条带强度对 RNA 的表达进行定量(一种半定量方法)。例如,对起始 cDNA 进行连续稀释并扩增。通过凝胶电泳使不同起始量的终点 PCR 得率可视化(图 2),然后对条带强度进行定量,并以管家基因为参照进行标准化,预估扩
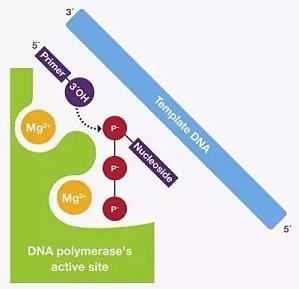
开篇之前先问个小问题您知道 PCR 的中文全称是什么吗?▼▼▼这是小编参加研究生面试时导师问的第一个问题,当时简直不敢相信自己的耳朵。如果您和我一样有片刻的犹豫,请备好小板凳,我们一起回顾一下简单 PCR 中那些需要我们了解的小知识。 PCR 基础知识PCR(Polymerase Chain Reaction),中文全称为聚合酶链式反应,是分子生物学研究中最为广泛应用的技术。在 70 年代的时候首次报道使用聚合酶及合成引物进行单链 DNA 的扩增,但直到 1983 年才正式作为一种研究工具用于 DNA 的扩增。自此以后,PCR 成为分子生物学研究不可缺少的一部分,被应用于从基础研究到疾病诊断、农业检测及法医调查等领域。其发明者 Kally Mullis 因此获得了 1993 年的诺贝尔化学奖。PCR 可在短时间内将单个 DNA 扩增得到成千上万个拷贝。其过程主要由 3 步组成:(1)变性,双链 DNA 经加热变成单链DNA;(2)退火,引物与模板的特异性结合;(3)延伸,DNA 聚合酶沿着模板,将引物的3’端延伸。 PCR 反应体系中的 6 大关键组分PCR 的成功取决于很多因素,其反

童鞋们都知道,要想成为真正的 PCR 达人,并非易事。一个小小的环节都可能让整个实验挂掉,也正是因此,PCR 成为了资深科研者们心中的痛。不要说实验本身,其实就连实验的耗材都隐藏着大学问,工欲善其事,必先利其器。今天我们就具体来说下 PCR 耗材中选择选择反应板的问题。PCR 板通常采用 96 孔和 384 孔的形式,其次为 24 孔和 48 孔。使用的 PCR 仪和正在进行的应用的性质将决定 PCR 板是否适合您的实验。裙边PCR 板的“裙”是板周围的板。裙边可为反应体系构建时的移液过程提供较好的稳定性,以及在进行自动机械处理时提供更好的机械强度。PCR 平板可分为无裙边,半裙边和全裙边。无裙边板缺少周围的面板(图 A)。这种形式的反应板可与绝大多数 PCR 仪和实时 PCR 仪的模块适配,但不适用于自动化应用。半裙边板在板的边缘周围具有短的边缘(图 B),在移液过程中提供足够的支撑,Applied Biosystems PCR 仪大多数均采用半裙边板。全裙边的 PCR 板具有覆盖板高度的边缘面板(图 C)。这种板形式适用于具有突出模块的 PCR 仪(可有利于自动化操作),可安全稳固
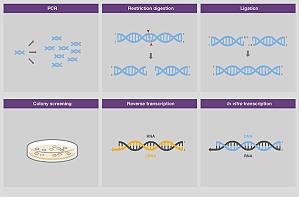
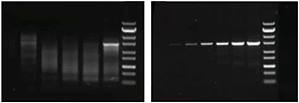
核酸电泳在许多分子生物学研究应用中都可用来检验实验结果,有时也可用于分离和纯化样品,以便进行后续应用。因此,常规核酸电泳的应用通常可以分为分析和制备两种类型,两者均依赖于分离、溶解和定量技术。 1.用于确认实验结果的分析型电泳在进入下一步工作流程或另一组实验之前,可使用分析型核酸电泳检验实验结果。正如下文所述,该方法主要针对凝胶中是否存在目标条带,以及条带的强度、迁移模式、迁移率和杂交情况进行评估。a. 检验酶促合成、消化和克隆实验是否成功核酸的电泳分析通常在以下技术(图 1)后立即进行以确定实验的成功和效率: 在几个小时内,聚合酶链式反应或 PCR 将目标序列的一个拷贝放大到数百万份拷贝。在终点 PCR 之后进行电泳,以确认靶标的扩增及其产量。在与限制酶反应以切割 DNA 底物上特定序列的限制消化反应后,在凝胶上运行的样品来确定 DNA 切割模式(以及消化完成的程度)。在分子克隆中,通过称为 连接的过程将 DNA 片段插入载体中。在一些情况下,可以在连接后进行电泳以评估反应效率(图 2)。然后将连接产物用于转化克隆生物体的 感受态细胞,如 大肠杆菌繁殖,然后筛选形成的菌落以确定它们是

凝胶电泳是许多分子生物学实验的重要部分。建立核酸电泳需采取一系列步骤来实现核酸样品的最佳分离和分析。核酸凝胶电泳工作流程1、选择和制备凝胶琼脂糖和聚丙烯酰胺是核酸分离中最常用的两种凝胶基质。两种材料都是三维基质,孔径大小适合核酸分离,且与样品间无反应。可通过改变基质的百分比来调整孔径大小,从而有效分离不同大小的核酸。有关琼脂糖凝胶和聚丙烯酰胺之间的选择,主要取决于核酸样品的大小和所希望达到的分辨率,虽然凝胶灌制和样品回收的方法也可考虑(表 1)。琼脂糖凝胶的孔径大小非常理想,可分离 0.1-25 kb 范围内的核酸分子。聚丙烯酰胺形成的孔径较小,可用于分离小于1 kb 的核酸分子。某些情况下,可采用聚丙烯酰胺凝胶以获得片段小于 100 bp 的单碱基分辨率[1]。表 1. 琼脂糖凝胶和聚丙烯酰胺凝胶之间的差异2. 准备标准品和样品a .核酸标准品选择当运行凝胶时,含有已知大小的核酸参照样品通常被称为标准品、标记或分子量标准,用于目的样品大小的估计。在为给定样品选择合适的分子量标准时,需考虑如下因素:Ladde 类型(例如 DNA 或 RNA),片段结构(例如单链或双链),构象(例如超螺

《分子克隆》虽然伟大的、人手必备的“分子生物学宝典”——《分子克隆》——里涵盖了分子生物学领域几乎各种基础技术(新中文版三册足足 3.5 公斤,知识就这么沉重),但实际咱们平时说的分子克隆技术主要是指将含有“目的基因的 DNA 片段”以“体外重组技术”插入某种 DNA 载体上,得到一个重组 DNA 分子。分子克隆技术现在早已算不上是“超酷”的前沿技术,只能算分子生物学基础入门技术,但它依然是整个分子生物学基础的核心之一,熟练掌握分子克隆技术之关键、又快又准地完成分子克隆依然是实验必需的看家本领。经典分子克隆技术vs无缝组装克隆技术技术总是随着时间向前发展的,由难到易,化繁为简,将不能变为可能。分子克隆技术的发展完美体现了这一点。 以构建表达载体为例,经典分子克隆中,要将目标片段插入载体,全靠内切酶的拆分和连接酶的拼接。关键是选择合适的酶切位点,使得酶切“拆”得的目标片段末端和相应载体末端正好能互补“拼”在一起,连接酶才能连起来。选择酶时首先不能破坏目标基因完整性(包含从起始密码到终止密码)及载体完整性;其次,尽量避免选两个酶切生成粘端一样的(粘端互补)——两边粘端一样的载体“偏爱”自我
上篇我们对经典分子克隆技术和无缝组装克隆技术进行了比较,经典分子克隆兜兜转转、如琢如磨,而无缝组装克隆技术却能做到操作简单和方便快捷。那它究竟是如何实现的呢?本期我们就来具体聊聊无缝克隆的四步致胜奇招。无缝组装克隆之 Step 123——带你飞,把还在用传统方法“琢磨”的他甩在后面基于同源序列的无缝组装克隆技术,关键是利用 PCR 引物,在目标片段两端引入与线性化载体两端同源的一段序列。基本策略是选定插入外源片段的位置,以此线性化载体设计包含线性化载体两端序列和目标片段两端序列的引物,扩增目标片段扩增片段与线性化载体、组装预混酶混合反应转化感受态细胞 · Step 1 · 如果载体上正好有合适插入目标片段的酶切位点,酶切载体使之线性化。为了降低单酶切载体不完全的几率,减少载体自连和提高筛选成功率,强烈建议双酶切——后面一个切点只要距离第一个切点 5 个碱基以上就行(主要是给两个酶留出结合空间)。如果没有合适的酶切位点,根据选定插入位点设计引物,用反向 PCR 来获得线性化载体。在多片段组装克隆的世界里,凡是酶切搞不定的,统统 PCR 引物设计搞定。简单粗暴有效。 · Step 2 ·
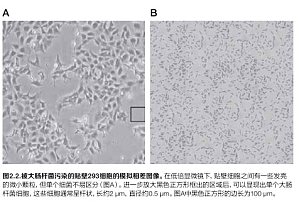
细胞培养物污染往往是细胞培养实验室中最常见的问题,有时会造成非常严重的后果。细胞培养污染物可分为两大类,一类是化学污染物,如培养基、血清和水中的杂质,包括内毒素、增塑剂和洗涤剂,另一类是生物污染物,如细菌、霉菌、酵母、病毒和支原体,以及其他细胞系的交叉污染。虽然污染无法完全消除,但可以通过全面了解其来源并遵循良好的无菌技术来降低污染的发生频率和严重性。本文将概述主要的生物污染类型。细菌细菌是一大类普遍存在的单细胞微生物。细菌的直径通常只有几微米,其形状多样,如球状、杆状和螺旋状等。由于分布广泛、生长迅速和体积大小等特点,细菌以及酵母和霉菌是细胞培养中最常见的生物污染物。细菌污染在培养物感染后几天内就很容易被肉眼观察到;受感染的培养物通常会变得浑浊,有时表面会有一层薄膜。经常还会出现培养基的 pH 值突然下降的情况。在低倍显微镜下,细菌以细小颗粒的形式出现在细胞之间,在高倍显微镜下观察可以分辨出单个细菌的形状。下面的模拟图像显示了贴壁培养的 293 细胞被大肠杆菌污染。酵母酵母是真菌界中的单细胞真核微生物,大小从几微米(常见)到 40 µm(罕见)不等。与细菌污染一样,被酵母污染的培养物

商品化试剂和培养基均经过严格的质量控制以保证其无菌,但它们在操作过程中可能被污染。请遵循以下指导原则进行无菌操作,避免污染。请始终使用适当的灭菌方法(如高压灭菌器、除菌过滤器)对实验室中配制的任何试剂、培养基或溶液进行灭菌。无菌操作请始终用 70% 乙醇擦拭双手和工作区。将容器、培养瓶、培养板和培养皿放入细胞培养通风橱之前,先用 70% 乙醇擦拭其外部。不要直接从试剂瓶或培养瓶中倾倒培养基和试剂。使用无菌玻璃或一次性塑料移液管和移液器操作液体时,每只移液管只能使用一次,以避免交叉污染。无菌移液管到使用时才能打开其包装。移液管应存放在工作区内。试剂瓶和培养瓶使用后应盖上盖子,多孔板应用胶带密封或放入可重复密封的袋中,以防止微生物和悬浮污染物进入。无菌培养瓶、试剂瓶和培养皿等到使用时才能打开盖子,不得将其开放暴露在环境中。使用后,应立即盖好盖子。盖子取下后,必须开口朝下放置在工作台面上。必须使用无菌的玻璃器皿和其他设备。进行无菌操作时,不要交谈、唱歌或吹口哨。尽可能快速地进行实验,以降低污染风险。 文章来源:赛默飞世尔科技
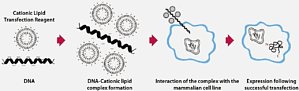
转染是将核酸导入真核细胞中的过程,是细胞生物学、基因表达和基因抑制实验中的关键步骤。转染是采用除病毒感染外的其他方法将核酸(DNA 或 RNA)人工导入细胞的过程。采用各种化学、生物学或物理方法导入外源性核酸会改变细胞的特性,从而实现细胞基因功能和蛋白质表达研究。转染后,导入的核酸可以瞬时性地存在于细胞内,只表达一段时间且不会复制,也可以稳定地整合至受体基因组内,随着宿主基因组的复制而复制。转染的目的转染的两个主要目的是生成重组蛋白,或特异性地提升或抑制转染细胞中的基因表达。因此,转染是一种功能强大的分析工具,可用于基因或基因产物的功能和调控研究,用于生成转基因生物,并用作基因治疗方法。基因表达转染最常通过使用质粒载体或 mRNA 在培养细胞(或动物模型)中表达目的蛋白。利用真核细胞中的蛋白表达可以生成经过适当折叠和翻译后修饰的重组蛋白。另外,将带有可检测的标记物及其他修饰的蛋白质导入细胞,可用于启动子和增强子序列或蛋白: 蛋白相互作用的研究。此外,根据转染策略的不同,转染还可应用于各种形式的生物生产。例如,导入重编程转录因子可以生成诱导多能性干细胞(iPSC)。另一方面,稳定转染提供

原创 SOOF 生物学霸 2022-04-20 17:594 月 12 日,江苏集萃药康生物科技股份有限公司开启申购,在公司公开的招股书中显示该公司主营业务为实验动物小鼠模型的研发、生产、销售及提供相关技术服务。部分品系的小白鼠单价高达万元以上,部分品系毛利率更是高达95% 左右。 随后一条「如何看待南大教授靠卖基因敲除小鼠,一只11723元,年赚近4亿」的讨论登上知乎热榜。 图片来源:知乎 作为天坑专业之首的生物,一直以来在大家的印象里总是和辛苦,清贫,回报周期长的标签挂钩。「教授靠卖小鼠,一年狂揽近四亿」的消息,给科研民工们带来的冲击力无疑是巨大的。 这究竟是不是暴利?在实验室里与小鼠为伍的学霸菌擦干贫穷的眼泪给大家细细道来 小鼠真香:狭窄赛道诞生两家上市公司 2017 年,南京大学的长江学者高翔教授成立了一家名叫集萃药康的公司,专业从事实验动物小鼠模型的研发、生产、销售及相关技术服务,他自任董事长。短短四年,这家卖小鼠的公司合计营收近 9 亿。 图片来源:公司招股书 无独有偶,另一家同样提供实验小鼠的公司南模生物也有着不错的销售业绩。2020 年,在公司成立 20 年时营收为
他们身处丰饶之中,却逐渐饥饿至死。」—— 阿瑟·克拉克《2001:太空漫游》 近日,北京市朝阳区公布了 2022 年公考拟录用人员名单,一时间引发热议。 此次拟录用的单位涉及部分政府部门及街道,设置的岗位包括城管、党群工作岗位等,拟录用 208 人。 纵观这份名单中的「学历」和「毕业院校」两栏,大半以上的拟录取人员都是国内 985、211 高校以及海外名校毕业的硕士,更是有三位顶尖名校的博士生赫然现身其中,挑动了大家敏感的神经,本科生反而成了「少数群体」。 这也难怪此事会引起议论纷纷——若是遮住这份名单的标题,又有谁会想到,毕业于外交学院、中国社科院大学的硕士争相报考的职位,其实是月薪约 4.9K 的「城管队员」呢? 所以其实也不难想象,当「北大博士考街道办城管」这样的话题冲上各大社交平台热搜榜时,网友纷纷表示:「宇宙的尽头是编制」,「比博士更高的学位是编制」。 图片来源:微博 不过,无论是支持或反对的论调,无非都代表着自己背后的一套行为逻辑;而做出选择的硕博们本人,心中也总是各有各的理由和打算。 我们没必要去为这些属于个人的选择和看法分个高下对错,但也正是在

DimAb®重组单克隆抗体开发平台缔码生物拥有独创的DimAb®重组单克隆抗体开发平台,可以在完成动物免疫后直接从外周血中获取阳性B细胞并克隆抗体基因序列。从单只免疫动物中我们可以得到上千个靶点结合阳性B细胞克隆,从中我们能够更为高效的筛选出具有高特异性,高灵敏度,和高亲和力重组单克隆抗体。1. DimAb®平台流程2.DimAb®平台优势3. DimAb®与其它抗体开发平台比较 B细胞抗体开发平台可成药性更高一个新开发的 ...
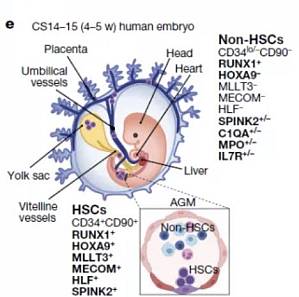
造血干细胞(haematopoietic stem cells, HSC)具有无限复制自身并分化成人体内各种血细胞的能力。几十年来,医生们一直在用捐献者骨髓和新生儿脐带内的造血干细胞进行移植,以治疗血液病和免疫系统疾病。然而,由于缺乏匹配的供体,且脐带血中干细胞数量较少,这些治疗也受到了限制。 研究人员试图利用人类多能干细胞在体外诱导生成造血干细胞,从而克服供体的局限性。然而,目前却依然无法实现,部分原因在于现阶段科学家只能将实验室培养的细胞分化成短寿命的造血祖细胞,而不是具有自我更新能力的造血干细胞。 然而,人类 HSC 产生的具体步骤尚不明确,因此也限制了从人类多能干细胞(hPSCs)诱导分化为能够移植的 HSC,同时限制了某些疾病模型的建立和相关研究的开展。 2022 年 4 月 13 日,来自加州大学洛杉矶分校的研究团队创建了一份史无前例的造血干细胞发育路线图,追踪了人类胚胎中造血干细胞发育的每一步,为人们在实验室中生产功能齐全的造血干细胞提供了蓝图。文章以 Mapping human haematopoietic stem cells from haemogenic endo
清明节刚刚过去,陶丰就等来了坏消息。 这位堪萨斯大学(University of Kansas)化学工程与化学系的前教授,在 4 月 7 日的裁决中,被判犯有三项电信诈欺罪和一项虚假陈述罪,可能会面临数十年的监禁,以及每项罪名高达 25 万美元的罚款。 图片来源:endpointsnews 受到如此指控和量刑,不仅因为陶丰的身份是华裔,而且与他「隐瞒与中国的关系」有关——他疑似曾与中国某大学签订了一份为期五年的合同。 以上这些判决和所谓的「依据」,都源自于美国政府从 2018 年展开的「中国行动计划」(China Initiative)。陶丰作为这一计划实施后被起诉的第一名被告,此前就已遭到逮捕,失去人身自由已 3 年——堪萨斯大学停止了他副教授之职,并且禁止他进入学校,还要求他在脚踝上佩戴追踪器。 据《麻省理工科技评论》统计,截至 2021 年,该计划总共起诉了 150 人,这些遭到政府迫害的人里,有将近九成都

G 蛋白偶联受体(GPCR)作为目前已知的人类基因组中最大的膜蛋白家族,可谓是闪闪发光的「明星蛋白」,其负责 80% 左右的跨膜信号转导,参与调控人体中大多数病理与生理过程。据统计,目前世界药物市场上至少有三分之一的小分子药物是 GPCR 的激活剂或者拮抗剂,掌握了 GPCR 的精细结构便是获得了生命密码! 2012 年,诺贝尔化学奖众望所归地花落 G 蛋白偶联受体研究领域,两位美国科学家罗伯特·莱夫科维茨(Robert J. Lefkowitz)和布莱恩·克比尔卡(Brian K. Kobilka)因在「G 蛋白偶联受体研究」领域的开创性卓越贡献,获得了顶尖褒奖,其杰出工作令人振奋与敬佩。 2022 年 4 月 13 日,四篇解析 GPCR 前生今世之谜的 Nature 论文同日齐发,其中三篇论文出自我国科学家之手。江山代有才人出,我国的科研工作者们亦数十年如一日地在 G 蛋白偶联受体(GPCR)研究领域深耕,做出了不起的成果! 图 1:来源 Nature

