一篇搞定 IP、co-IP 实验
赛默飞世尔科技
某个实验间隙…
萌新:开题报告已提交,转眼 1 个月过去了,实验还是一头雾水,为啥我的 co-IP 拉不下来蛋白?
奋斗中的师兄:我当初正好相反,拉下来的蛋白被抗体条带覆盖住了,换个抗体忒不好找,无奈换了个蛋白,还好挺顺利。
大师兄:蛋白吧,不但种类多,理化性质差异较大,关键它们又喜欢组队在细胞里干活,IP、co-IP 又是常用的手段,掌握好这个工具还是很重要的,总不能随随便便就换个蛋白。
那要怎么快速掌握这项实验,轻松搞定各种疑难杂症呢?
我们依据三十多年的 IP/co-IP 实验经验,总结了一线技术支持常被问到的问题,希望这篇纯干货能帮助大家顺利升级打怪,早日驾驭 IP、co-IP。
IP、co-IP、pull-down 简介
IP(Immunoprecipitation),免疫沉淀:是利用固定在磁珠或琼脂糖树脂等基质上的特异性抗体对抗原进行小型亲和纯化的一种方法

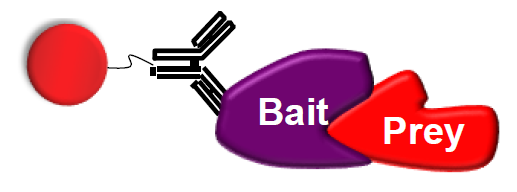
Co-IP(co-Immunoprecipitation),免疫共沉淀: co-IP 是一种常用的鉴定蛋白-蛋白相互作用的方法,其基本实验流程和IP类似,通过使用诱饵蛋白(IP 中的抗原,bait protein)特异性抗体间接捕获与其结合的靶蛋白(prey protein)。
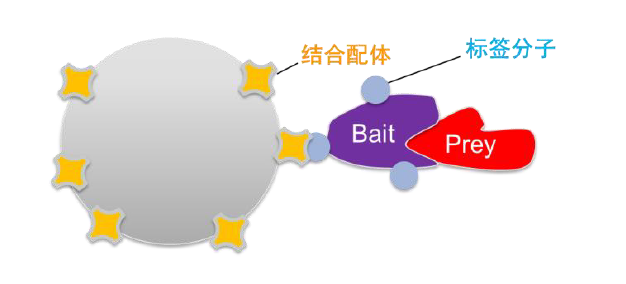
Pull-down:另外一种进行蛋白-蛋白相互作用研究的方法,针对无法找到可用的抗体的靶蛋白。可以采用一个表位标记蛋白质(可在细胞内组成型表达),将其固定在基质上获得互作蛋白。很多蛋白质由于无法找到可用的抗体而无法进行免疫沉淀。
co-IP 实验流程
由于co-IP 相对问题较多,本文以 co-IP 为例进行介绍
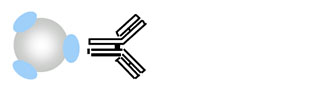
Step 1:固定抗体
Step 2:加样

Step 3:结合,去除非特异性蛋白
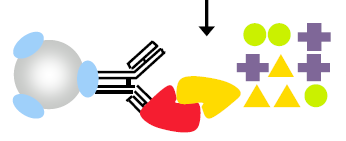
Step 4:靶蛋白(红色)洗脱或者互作蛋白复合物(红色+黄色)洗脱

Step 5:分析检测—Western Blot 或者质谱
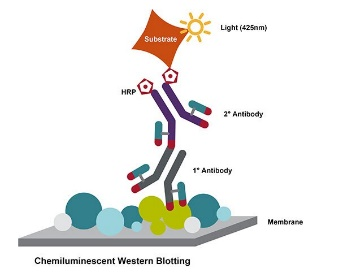
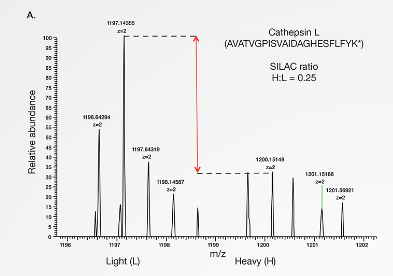
常用“黑话”

以上示例图证明了 EDR1 和 EDR4 有相互作用。如果想要设计好实验,先清楚以下常用“黑话”含义。
阳性对照:证实细胞裂解物中确实存在靶蛋白(IP)或者互作蛋白对(co-IP,比如诱饵蛋白X和靶蛋白 Y),即为常说的 input。可直接取抽好的蛋白溶液进行 Western。
阴性对照:用来排除假阳性。IP/co-IP 后靶蛋白或者互作蛋白对都检测到了,固然是值得高兴的,但是这结果是不是假阳性呢?还得做个阴性对照。阴性对照如何设置呢?考虑到整个实验体系会出现 beads、抗X抗体、诱饵蛋白 X、靶蛋白 Y(co-IP):
|
可能出现的假阳性来源 |
需要的阴性对照 |
|
抗 X 抗体结合非特异的蛋白 |
与 IP 一抗种属亚型相同的免疫球蛋白(比如,抗 X 抗体是 Mouse IgG,阴性对照可使用 Mouse 免疫球蛋白) |
|
beads 对蛋白的非特异结合~K~Hstrong~M~K~Hp~M~1~K~Htd~M~1~Ktd~M~1~Kp~M同上,此外还可进行 Pre clear,IP 正式实验前排除非特异结合蛋白。如果使用琼脂糖,建议进行 Pre-clear;如果使用磁珠,此步可忽略 |
|
|
抗 X 抗体的轻链(25 kD)和重链(50 kD) |
更侧重于如何在实验上进行优化(见下文) |
pre-clear:上样前先用裂解液过一遍柱子,减少基质本身对蛋白的吸附
Output:在某些 co-IP 实验中,实验人员会把IP后的上清分别进行诱饵蛋白 X 和靶蛋白 Y 的 WB 检测,该对照组称为output 组。
内源性 co-IP 实验,如果最终结果为阳性,则可以证明两个蛋白之间存在相互作用;但是结果为阴性,是否两个蛋白就一定不发生相互作用了了呢?也不一定。有可能是两个蛋白是弱相互作用力,这时候可能需要交联剂协助,锁定强化弱互作蛋白;也有可能是内源蛋白表达量低,可先做过表达 co-IP 作为对照。
实验前的思考
1、抗体----选经过 IP 验证的抗体,减少假阳性概率
2、抗体结合蛋白---Protein A, Protein G, Protein A/G, Protein L?
- Protein A—只与 Fc 区域结合
- Protein G—结合 Fc 区域和部分轻链
- Protein A/G—重组蛋白,兼具 Protein A 和 Protein G 的结合位点
- Protein L—结合仅限于那些含有 κ 轻链的抗体
省事且通用可选择 Protein A/G,也可根据结合力表格进行选择,详见资源信息部分
3、基质—琼脂糖还是磁珠?
|
琼脂糖 |
磁珠 |
|
|
优点 |
简单易用,无需辅助设备 |
操作简单 |
|
孔径大,直径大(50-150 um),结合力强 |
直径小(1-4 um),动力学好 |
|
|
靶蛋白获取量高 |
表面光滑,beads 吸附很少,背景低,抗体消耗少 |
|
|
缺点 |
多孔易吸附(记得做 pre-clear) |
需配备磁力架 |
|
其他 |
建议在 4 ℃孵育 4-6 h 或者过夜 |
依据不同品牌的磁珠类型,建议孵育时间 15 min-2 h |
4、孵育(结合)顺序与孵育条件
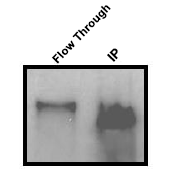 |
 |
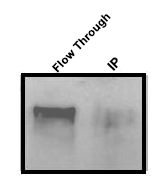 |
|
抗体+裂解液,然后和加入 beads |
Beads+抗体,然后加入裂解液 |
Beads+抗体+裂解液,三个一起上 |
从以上三个结果可以看到,第一种孵育方法抗原得率最高,第三种抗原得率最低。如果靶蛋白丰度较高,第一种和第二种差别并不显著。注意,整个体系选择同样的孵育顺序,以确保结果的重复性。
5、各种 buffer
整个 co-IP 系统,涉及到裂解液,结合液,洗涤液,洗脱液,这4种溶液要如何考量呢?
裂解液---样本质量很大程度受限于裂解液,要考虑其中的去垢剂种类。
- 非变性裂解液,含有非离子型去垢剂,有助于稳定天然蛋白构象,比如 IP lysis buffer。不建议使用变性裂解液,比如 RIPA,虽然裂解剧烈,但是无法维持蛋白天然构象,会破坏互作蛋白对。
- 如果自配,建议采用温和系统,比如 NP40/Triton 0.1%-1%;SDS ≤0.1%,盐离子≤150 mM
- 记得要加蛋白酶和/或磷酸酶抑制剂
结合液---利于蛋白之间的相互结合,要考虑孵育(结合)顺序
- 如果抗体和 beads 先孵育,建议 选择 pH 8.2(更常用)或者 pH 5.0
- 如果选用纯抗原作为诱饵蛋白 X 去拉裂解液中的靶蛋白 Y,抗体和抗原的结合 buffer 选择中性PBS/TBS
- 如果诱饵蛋白 X 和靶蛋白 Y 来自细胞裂解液,那么裂解液配方建议选择非变性裂解液,NaCl 浓度<150 nM,SDS 浓度<0.2%
洗涤液---去除非特异结合蛋白,顾及互作蛋白间的结合强度优化配方
- 如果互作蛋白作用较弱,建议选择 PBS 或者 TBS 进行洗涤
- 通常会加入去垢剂降低背景,比如 0.5-1% NP-40/TritonX-100/CHAPS
- 如果效果还不佳,还可增加盐离子浓度,比如<250 nM NaCl,以及加入1-2 nM DTT/β-ME
- 还可增加洗涤次数,比如 3-5 次
洗脱液---将目的蛋白从固相介质上洗脱下来,依据捕获方式和下游应用优化配方
- 如果诱饵蛋白或者靶蛋白带有标签,可用高浓度标签或配体进行竞争洗脱
- 温和的配方:0.1M 甘氨酸(pH2.5-3)
- 也可使用 SDS-PAGE 上样缓冲液直接洗脱
资源合集中有建议的对应不同洗脱环境的配方。
常见问题
1、Co-IP 背景较深或者条带杂乱
- 固相基质的非特异性结合—
- 抗体结合非特异的蛋白
- 洗涤不当造成杂带
- 建议方案:pre clear 或者更换磁珠;设置对照排除非特异性条带;优化缓冲液(学习视频中会有详细讲解)
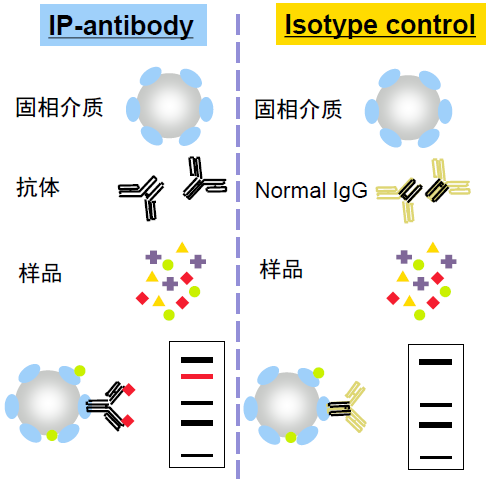
2、Input 条带清晰,co-IP 条带很弱?
设X为诱饵蛋白,Y 为靶蛋白,有可能为
- X 和 Y 的相互作用本身比较弱
- X 蛋白并不是 Y 的唯一,只部分 Y 与 X 互作
- 可以尝试反向拉,比如用 Y 去拉 X,可能效果比较好
3、我的 p 53 蛋白正好和抗体重链分子量接近,要如何减少抗体干扰?
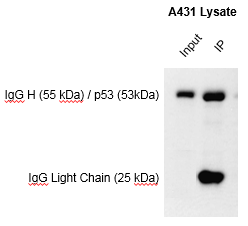
建议方案:
- IP/WB 一抗使用不同种属来源,选择与IP抗体种属无交叉反应的二抗(Cross Adsorbed secondary antibody)
- 特殊二抗:比如仅识别重链或者轻链的二抗,或者仅识别天然抗体的二抗
- 直接源头遏制:将抗体直接交联到 bead 上,或者将抗体交联到 protein A/G 上,避免洗脱干扰
4、靶蛋白没有条带
- 裂解液不合适,或者没有添加抑制剂,导致靶蛋白或者饵蛋白构象改变不能结合或者降解
- 饵蛋白较难洗脱,换成高强度的洗脱缓冲液
- 选用的抗体不合适,建议 IP 验证过的抗体,并优化下抗体用量
- 蛋白间为弱相互作用,或者相互作用依赖翻译后修饰
5、琼脂糖难离心,每次总会吸上来一点儿
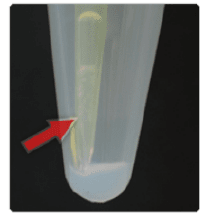
- 使用带滤膜的离心柱,下部有接样管,琼脂糖完全截留在滤膜上
- 换成磁珠
更多常见问题分析,详情请看学习视频
学习视频
由我们的技术支持精心准备的一系列免疫(共)沉淀在线课程,内容丰富详实,关于实验前思考和常见问题的分析更详细,还可随时观看。
目前更新的内容有:
《蛋白-蛋白互作常见研究方法介绍》
《免疫共沉淀实验详解》
《免疫共沉淀常见问题及解决方案》
文章来源:赛默飞世尔科技








