




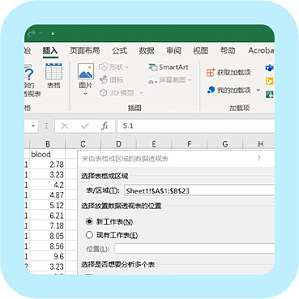
作为医学生,搞科研常常离不开调查问卷。然而发问卷一时爽,数据分析火葬场。来自问卷的数据乱七八糟,分析该从何下手真是让人苦恼。问卷数据分析有技巧,分析前的准备不可少!SPSS 是科研数据分析的常用软件,科研狗几乎人手一个。那么,基于 SPSS 软件的问卷分析,需要做哪些准备呢? 常见问卷题目类型:问卷题目的常见类型有填空题、选择题和排序题。☆ 填空题在问题后面的横线上或括号内填写答案。例如:您有_____个兄弟姐妹。☆ 选择题包括单项选择题和多项选择题。例如:单选题:您的性别是:(1)男 (2)女多选题:您通过哪些方式学习 SPSS?(可多选)(1)课堂学习 (2)自学 (3)公众号学习 (4)腾讯课堂 (5)B 站 (6)其他☆ 排序题在列举的选项中按顺序依次排序。例如: 您认为疫情期间配备防疫物资的次序为: ①护目镜_____ ②防护服_____ ③手套_____ ④鞋套_____ ⑤口罩_____ 一、原始问卷数据导出导出数据有两种类型:(1)用数字 1、2、3、4 等代表选项 1、2、3、4;
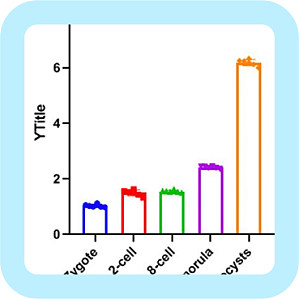
近期还不能开学,学习就只能在家里进行了,偶然在课题组群里听到师妹在讨论荧光定量结果的统计分析。她们在那里「瞎折腾」,也没有想到去请教一下师兄师姐。就两组数据,先下载了一个 SPSS 软件,整半天还整不明白怎么分析。查阅了各种教程与书籍,终于分析好了,结果又「傻眼了」,组图又成了一个问题。作为一个有经验的师兄,很是心痛啊!就这么简单的数据用 SPSS 分析,本身就是大材小用,「杀鸡用牛刀」。师妹,用 GraphPad prism 吧,这个软件作图与分析是一体化的。为避免更多的师弟师妹走弯路,今天我们就来说说 GraphPad prism 。软件可在官网下载:https://www.graphpad.com/scientific-software/prism/ 一、基本分析与作图首先,从师妹的问题入手,我们说一下这个软件的基本分析与作图。数据结构如下(已经处理好的表达量情况),探究一个基因在小鼠胚胎发育早期过程中的表达变化。图片来源:电脑截图打开 GraphPad 软件,GraphPad 特别人性化,提供多种作图模式,基本上满足了我们实验数据分析作图中的方方面面,小伙伴可以根据不同的需求选
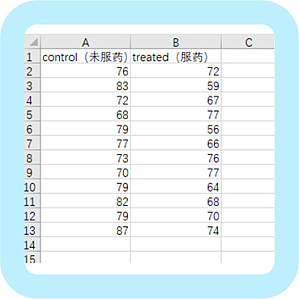
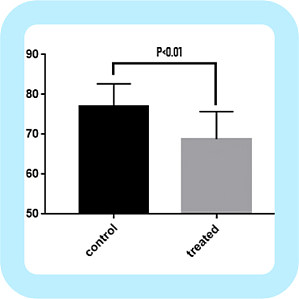
统计作图对很多医学工作者来说是一件非常头疼的事,虽然本科一般会开课教学,但那时估计大家都是抱着学数学的想法,及格就万事大吉,意识不到它的重要性,直到开始做科研、写文章才知道,统计作图会一直伴随着你!统计作图软件有很多,简单的作图可以用 excel 完成,但你若想绘制稍微美观和复杂的图形,就会发现 excel 真的很笨。也有 R 这种大神绘图软件,只有你想不到,没有它画不出的!但是,像小编这种统计业余选手,整整学了好几个月才参透一二,应用起来还是十分困难。说了这么多,靠谱的小编要来给大家安利啦!又能统计分析,又能作图,操作简便,简单粗暴即可绘制大部分医学 SCI 统计图的软件——GraphPad Prism。不多说,干货来了,以一个简单的临床数据为例,给大家展示一遍 GraphPad 的作图方法,看了就知道有多人性化!举个例子:这是两组心律失常患者,运动恢复后心律数据,B 组在运动前服用抗心律失常药物,现要分析两组患者运动后心律指标是否有差异。(数据如图)开始作图,首先打开软件。在新建窗口中绘制图形选项选择「column」,数据类型为第一个独立样本,点击「create」。然后复制数据到

对于相关和回归的关系,教材是这么说的:相关用于说明两变量之间的关系方向和密切程度,没有主次之分;回归更进一步用于定量刻画两变量在数值上的依存关系,可以依据专业拟定主次。我们在学习和工作中还常见下面这些表述:(1)相关是回归的基础,无相关就无回归。(2)相关程度越高,回归方程的拟合程度就越好。(3)能进行回归分析的变量之间存在相关关系。(4)相关是一种双向变化关系,回归是一种单向变化关系。(5)对于新数据,可先做散点图,求出相关系数,对于确有相关关系的变量再进行回归分析。(6)相关系数(r)和回归系数(β)的方向一致,可以相互推算。(7)研究两个变量的相互关系用相关分析,研究两个变量的依存关系用回归分析。正因为教材中的定义和平时常见的表述,在实际应用中两者容易混淆,对于自己的数据,不知道是该用相关,还是该用回归,或者该用哪种回归。 问题一:没有相关关系就不能做回归分析吗?我们知道在回归分析之前,首先需要了解变数间是何种相关关系,才能选择适当的回归模型。但大千世界关系复杂,看似无序的两者,可能存在某种特定关系,因此很多时候看似无序的数据,经过分层、分组、多因素或合适的模型处理,才能发现有意

数据统计分析是一个让大家头痛的问题,统计方法之多,以致于拿到数据后往往都无从下手。临床研究对统计分析的要求尤其高,也是审稿人的关注点。因此要做好临床研究,首先要掌握统计分析。1. 分析数据类型及分布特点每种统计方法对数据类型及分布是有要求的,这是我们在选择正确统计方法前必须要考虑的。对于来自于临床样本的数据和实验结果,由于个体差异大,绝大部分连续性变量都不符合正态分布,除非是大数据(如正常人群白细胞数,血糖等这些数据会是正态分布)。因此,今天笔者主要介绍非正态分布的连续性变量的表示及组间差异的统计分析。对于数据分布特点(正态分布和非正态分布)可以通过直方图分布特点判断或者 D 检验、W 检验来判断。下面就这两种方法举例分析(SPSS 分析)。P>0.05 为正态分布,反之为非正态分布。从直方图也可以直观的判断,Group1 为正态分布,Group2 为非正态分布。图片来源:软件截图 2. 集中和离散趋势的表示对于非正态分布的一组连续性变量,选用均数(Mean)和标准差(SD)表示是不准确的(很多初学者,刚开始写文章,大多选用的是这种表示方式)。一般选用中位数 (Median) 和
一、忽略了样本纳入量的计算和说明对于一个研究而言,一般而言先要计算样本纳入量,否则做出来的研究极有可能没意义。笔者曾经接到过一位麻醉学博士的求助电话,叙述他在预答辩环节,被问「为什么你的研究 A、B 两组选择这么多例老鼠」时,他答不上来的焦虑。而当这个问题被临床流行病学方面的专家问到时,基本上文章也就被判「死刑」了。这个文章就属于具有严重问题——没有说明样本纳入量的问题。 二、 明显的造假行为科学研究一定要严谨,一定要杜绝学术造假。笔者曾看过一篇文章初稿写着:将患者通过中央随机系统分为 A、B、C 三组,A 组 30 例,B 组 30 例,C 组 65 例。当时这篇文章被怀疑涉嫌数据造假,因为很少随机分组能分出 3 组相差这么多的。随后怀疑被证实,我们要求作者解释为何会出现这种情况时,他送回来的修改稿将 A、B、C 三组的例数改成了 60,60,65 例。当然这篇稿子秒退,理由(高度怀疑数据造假)。 三、对「随机」概念理解有误对于某些基本的概念理解有误,最常见的问题集中在「随机上」,很多临床的作者把随意法和交替制定法认为是「随机化」,而一个不恰当的随机,可能造成选择偏倚和混杂偏倚的渗
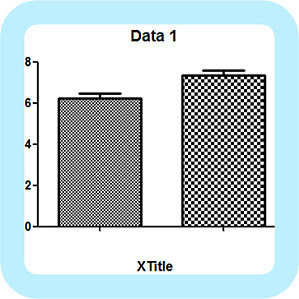
很多搞科研的小伙伴习惯用 GraphPad Prism 软件作图,殊不知在分析数据方面,GraphPad Prism 也是一把好手。今天,笔者就分享一下用 GraphPad Prism 分析数据的方法。 第一部分:利用 GraphPad Prism 分析数据的步骤1. Analyze 对话框在学习具体步骤前,我们首先要知道 Analyze 的功能,它不仅包含了统计和回归,还能对数据进行处理,例如转换、删除基线和标准化。接下来,我们以比较 A、B 这两组数据为例:图片来源:作者本人制作将两组数据输入软件用于记录数据的位置后,点击「Analyze」。需要注意的是,无论你选择全部还是部分数据,GraphPad Prism 都会对所有的数据进行分析。图片来源:软件截图点击后,会出现如下的对话框,这时候弹出对话框的左上角会默认 Built-in analysis,我们不要去更改它。图片来源:软件截图对话框左侧,为各种分析方式,我们根据我们需要的分析类型进行选择即可。分析类型选项通常根据它们关联的数据表类型进行分组,你可以更改为自己想要的分析类型。接下来,在对话框右侧选择数据组进行分析,默认情况下
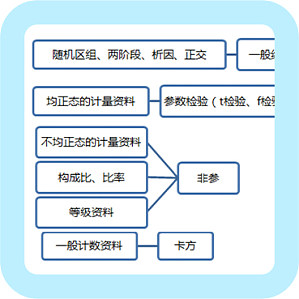
在我们处理实验数据的时候,最常用的统计学软件就是 SPSS。SPSS(Statistical Product and Service Solutions)「统计产品与服务解决方案软件」,是用于统计学分析运算、数据挖掘、预测分析和决策支持任务的软件产品及相关服务的总称。 但是我们学习 SPSS 的重点并不在学习软件本身,而是学习相关的统计学知识。今天笔者根据多年的理论学习和实操经验,让各位科研者突破 SPSS 统计学理论,快速掌握 SPSS。 SPSS 是一个半傻瓜式操作软件,只要认识了软件基本界面和功能,然后把你准备好的数据输进去,点击需要进行分析的功能,软件会自动给你算出分析结果,并不需要写代码或程序。所以真正难的是输入数据这一步。 我们首先了解基本统计知识,接着选择正确的检验方法进行结果解读;与此同时,掌握一些数据清理的技能更好,比如对异常数据处理。一、基础知识(一)四种指标类型1. 两种基本指标类型图片来源:自己做的2. 两种特殊指标类型 等级资料是特殊的计数资料,跟一般的计数资料不同的是具有方向,比如:+、++、+++(用非参

年底将至,每次进实验室都能听到大家纷纷惨叫:「怎么又没做出结果!」「什么时候才能凑齐一张 figure?」「明年评职称又没戏了!」......小编想说,有时间有基金做实验还算不错,如果碰到老板说「课题随便做,只要不花钱就行」,那如果你想通过做实验发文章可真的要 go dead 了。当然,善良的小编怎么会让你轻易放弃 SCI 呢,给你指明另一条路吧,做生信!今天给大家推荐一款高级软件:RR 语言作为一款免费的开源软件,可自定义语法,具有高度可扩展性和代码开源性,已成为众多医学工作者分析临床大数据和医学大数据的首选软件。但许多人仍然面临着想做大数据分析却不会 R 语言操作的困境,于是对大数据分析望而却步。虽然你之前可能没有编程经验,但是学习工作中经常需要计算、统计、绘图,那 R 是你的首选。语法结构简单,上手较快,而且函数和 packages 都有很好的实例文档。R 是一门自学型语言,来 R 吧,你不会孤独。 一、R 的下载安装小编亲自带领大家下载安装使用 R,超贴心哦!之所以可以这样,还是源于它是完全免费的。首先进入 R 的官网:【https://www.r-project.org/】按
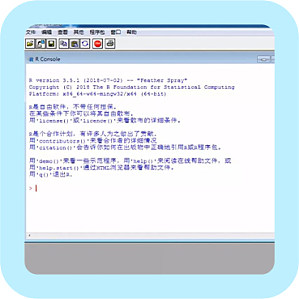
科研常用统计分析软件近年来市面上涌出一大批统计分析软件,每个软件都有独到之处,怎么选择合适的统计软件,对医生、医学生实属不易。哪些软件的统计结果权威,哪些软件专业绘图,哪些软件可对大数据综合预测分析?哪些软件一学就能会?又有哪些是免费的?今天,小编就来帮大家盘点几款医生、医学生常用的统计分析软件。 一、SPSSSPSS 这款老牌统计软件,可以算是统计软件的前辈了,最早是由美国斯坦福大学的三位研究员研发。与在国内广泛应用的其他统计软件相比,粉丝量(用户量)大,且不限于医学工作者,社会调研、心理学、教育学的很多统计工作都是由它完成。(偷偷告诉大家,小编的本科毕设统计图表全靠它呢)为什么它如此圈粉?优点:操作简单,页面友好,统计方法较为齐全。满足各行各业有统计学基本需求毫无压力,且容易上手。给大家看一下 SPSS 的界面吧,图一为设置变量界面,图二为数据录入界面: 数据录好后,就可用其强大的统计功能进行分析啦。缺点:非免费,而且价格较高。与 OFFICE 的接口不全,会造成很多不便之处,其次,软件语法运算还不够强大,结果呈现也不够清晰明了。 二、GraphPadGraphPad Prism
如何计算临床调查中样本量?统计老师说:针对不同的数据属性,可以根据已知的临床信息,采用相应的估算公式完成样本量估算或者借助相应的统计学软件完成这一过程。然而,他是统计老师。我们大部分人的统计都比较渣渣……图片来源:时景璞. 临床研究中样本量的估计方法 [J]. 中国组织工程研究, 2003, 7(10):1569-1571.碰到上图类似的公式几乎是「崩溃」的节奏!有次我们医院准备调研员工对新的绩效改革方案的满意度,院长大手一挥,每个人都填!!!所有人!!!可以看出我们院长的「豪气」,也能看出我们院长的统计学也很渣。这其实是临床调研中最极端的做法是搞全面调查 (普查),虽然没有了抽样误差,但是总误差可能比抽样调查更大。为什么这样说呢?因为普查涉及范围大,时间长,调查人员多,许多条件无法控制,因此,调查实施时就可能比较粗糙,在数据录入、处理时也更容易产生误差,特别是过大规模的抽样调查更容易发生类似的情况。而精心设计和实施的适当规模的抽样调查,则可以做得比较细致,数据处理时也比较容易控制误差。还有过大规模的调查必定耗时过长,待所收集的资料发表之时可能已失去其大部分价值了。临床科研者常见困惑但
无论是毕业答辩,还是研究设计,样本量的问题经常困扰着科研小白。样本量取多少合适?样本量是不是越大越好?临床上并没有那么多的样本怎么办?这些都是常见的临床研究疑惑。而样本量在计算的过程中涉及很多统计量、复杂的公式和专业的软件,让科研初学者望而却步。今天,给大家分享一个简单的样本量计算工具,助力科研设计(网址:http://powerandsamplesize.com/),网站中还提供了公式讲解和 R 语言代码等有关信息。进入网站后,可以通过点击红框内按钮直接进入样本量计算界面:图源:网站截图根据研究类型进入选定的计算模式后可发现界面可分为参数输入与结果输出部分、快速运算图、运算模式选择、计算公式讲解和 R 语言代码展示等若干区域。图源:网站截图计算样本量之前首先要明确研究类型,明确了研究类型以后就可以通过已发表的文献或前期预试验来确定关键结局指标、不同组别的人口学特征和试验药物疗效等相关信息。接下来,以等效性随机对照试验为例,简要讲解 powerandsamplesize.com 的操作方法: 例 1:某平行设计的随机对照试验拟探讨试验药物是否能降低收缩压。估计安慰剂对照组收缩压的平均值
震惊!作为一个科研工作者,最常用的软件竟然不是 SAS、SPSS、Graphpad、Origin 巴拉巴拉,而是 Excel、PS、AI?事实上,对于处理医学数据和图片来说,掌握一些奇奇怪怪的办公软件小技巧甚至比使用很多专业的软件更加方便快捷。比如,当你或者老板只是想看到基础的数据趋势,或者非常快捷地做一些组会的结果输出,Excel 就真的不要太快!接下来,各位小可爱就跟笔者一起回顾和学习一些比较常见的 Excel 偷懒(不是)小技巧吧! 一、入门款快捷键及便捷操作我们先回顾一下众所周知的一些 Office Excel 基础快捷键:1、Ctrl+S 保存键这个快捷键 Word、Excel、PPT 都能用到,务必吸烟刻肺牢记心中,每 5 分钟操作一次,导师再也不用担心我们会丢数据了。2、Ctrl+C 复制 / Ctrl+X 剪切 / Ctrl+V 粘贴再也不用右键复制、右键粘贴这么麻烦了。3、Ctrl+I做表格的时候一键斜体,灰常方便。 4、Ctrl+Z 一键撤销前一步操作,可以一直重复撤销,直到回归数据原样。5、Ctrl+A用来全选数据,配合复制、粘贴、剪切,左手右手一个快动作,转
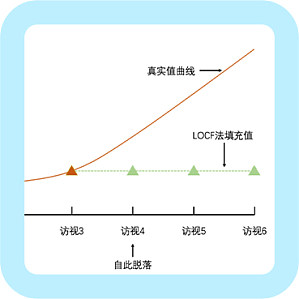
相信很多朋友都经历过临床数据缺失,看着一个个空白的格子束手无策。其实在临床试验中,病例脱落导致数据缺失是十分常见且难以避免的。在尽量避免不必要的数据缺失的同时,还应掌握了相应的缺失数据处理方法。 一、为什么会产生「缺失数据」?临床试验中的缺失数据一般是由于受试对象在试验中因依从性差、不良事件或疗效不佳等原因提前退出试验造成的;也可能是因为采集标本或测量过程中因样本量不足或灵敏度不够所造成的疗效指标缺失;也可能是在数据记录或整理过程中造成的数据丢失等。 二、缺失数据有哪些类型? 1. 完全随机缺失(missing completely at random,MCAR)是指缺失数据的产生完全独立于可观察到和不可观察到的数据,其发生的比例不会依赖于受试者的特点,理论上不会给试验结果带来偏倚,数据分析时可仅分析现存数据。例:因受试者搬家或出差等所造成的失访脱落。2. 随机缺失(missing at random,MAR)是指缺失数据的产生独立于不可观察到的数据,但会与可观察的数据有关。受试者在试验中因不良事件或因疗效不佳而脱落,而不良事件发生和疗效不佳并不是完全随机发生,可能与其他因素相关,这些
读研究生的同学,无论是硕士生还是博士生,都离不开科研实验。我们通过千辛万苦,得到了数据,这时候可不要以为万事大吉了,重头戏在后面呢——整理实验数据。今天,笔者就来分享一下自己整理数据的一点经验吧~ 一、「完整全面」地记录数据实验数据必须完整全面地记录,实验时间、实验步骤、实验结果缺一不可。好记性不如烂笔头。不要对自己的记忆过度自信,觉得记在心里就好了,等到准备写论文的时候面对一堆数据一脸茫然,这是什么,这又是什么...实验时间一定要写实验往往不是一次就可以都得到理想的结果,很可能需要重复,重复,再重复,这时候日期就非常重要了。遥想当年,笔者刚开始做实验的时候,有一个实验重复了 5、6 次,原始数据是 Excle 。当时,笔者对自己的记忆过分自信,觉得心里记住了就可以了,所以没有记录日期。结果几个月后想要写论文的时候,发现记忆早已经模糊,根本不记得哪个数据是自己所需要的,所以只能哭着把数据又全部分析了一遍……实验步骤一定要写作为实验狗,笔者做实验前,尤其是新实验(哪怕是实验室师兄师姐已经做了 N 遍的实验)先尽可能的写清楚实验步骤(Protocol)。能写多细写多细,细到别人照着可以完全

亲爱的护理伙伴们,作为一名科研路上的新手,你是否经常望统计学而生畏,觉得统计是你科研路上最大的拦路虎?但笔者想说,统计学在咱们护理科研中并不是最难的部分,与研究设计相比,统计学还是属于比较简单的。接下来,笔者就将自己学习统计学的经验分享给大家,希望对为此发愁的你有所帮助! 一、在文献阅读中学习文献阅读是科研写作的基石,平常咱们阅读的文献主要分为三大类:综述、横断面研究以及试验性研究。对于后两类文献,统计部分必不可少。通过长期阅读的积累,常用统计学方法有哪些并如何加以选择,你会逐渐了然于胸。在日常的文献阅读中,我们可以这样做:1、阅读专业领域的顶级期刊 顶级期刊的文章无论是研究设计还是统计学部分的描述都比较严谨,有很多地方值得我们去学习。图片来源:作者笔记截图笔者选取了发表在《中华护理教育》期刊上的《音频指导居家盆底肌锻炼对压力性尿失禁孕产妇产后盆底康复的效果评价》文章,通过介绍该文章中涉及的统计学方法,来具体讲解如何在阅读中学习统计学。接下来,笔者将通过如下示例进行介绍:图片来源:作者笔记截图图片来源::作者笔记截图图片来源::作者笔记截图这篇文章中的统计学方法主要有 t 检验、c2

「丁香实验」致力于帮助科研人轻松科研,在丁香实验小程序上线后,已经汇集上万条实验 protocol、常见操作问答、科研方法文章、线上直播课程等。在2022年下半年,我们希望能够更加深入校园,让更多的校科研er们,知道我们丁香实验,同时也希望给在校的科研er们带来一份不错的额外收入,所以校园兼职计划来啦~校园兼职要做什么 校园兼职是丁香实验在校园的代理人,主要工作内容为:完成丁香实验小程序的校园推广工作~你将会获得一份不错的现金回报和一场还不错的实践经验;ps、校园推广还处于新生阶段,我们期待你除了基础的推广工作,还能参与校园推广的持续优化。我们希望你1、身为科研人或者熟悉学校实验室科研的小伙伴们;2、充足的时间及开放的态度;3、了解及认可丁香实验;兼职报名方式扫码添加主编微信,说明兼职即可丁香实验小程序10000+实验 protocol
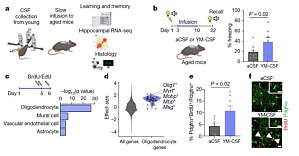
一、导读大脑老化是痴呆和神经退行性疾病的根源,给家庭和社会带来巨大负担。前期对模式生物的系统性研究表明适当的外界干预能够逆转包括大脑在内的多种组织的生物学衰退。例如,年轻血浆的输注可使老年大脑恢复活力并恢复记忆功能。然而,大脑受到了脑屏障的保护,这在一定程度上可能会限制这些干预措施的获取,进而阻碍它们的功能效应。 脑脊液(Cerebrospinal fluid, CSF)与脑细胞密切相关,它携带信号,指导发育过程中神经元祖细胞的增殖和特异性。然而,脑脊液蛋白组成会随着人类年龄的增长而变化,表现为炎症蛋白的增加和脑源性神经营养因子等生长因子的减少。不过,脑脊液中的这些变化是否与年龄相关的认知能力下降有关尚不清楚。 2022 年 5 月 11 日,来自斯坦福大学医学院神经学与神经科学系的科研团队在国际顶级期刊 Nature 发表了题为 Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17 的研究性文章,他们发现将年轻的脑脊液直接注入衰老的大脑可以明显改善记忆功能,其中少突胶质细胞对这种恢复最敏感,他们

南宋爱国诗人陆游曾言「我得宛丘平易法,只将食粥致神仙」,指出清淡饮食有助于延年益寿。早在一个世纪前,西方科学家发现通过降低卡路里的摄入能够显著地延长动物的寿命。延年益寿、青春永驻是人类对于生命的热爱的体现,是追求永生的美好愿望,引起无数科研工作者的研究兴趣。 2022 年 5 月 5 日,霍华德休斯医学研究所研究员 Joseph Takahashi 及其团队在 Science 杂志发表研究论文 Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice。 这项最新的研究表明,在正常能量摄入基础上减少 30%~40% 的卡路里限制饮食可以让小鼠的寿命延长 10%。但如果将进食时间只限制在夜间(小鼠最活跃的时间),可大大延长低热量饮食小鼠的寿命,整体寿命延长了 35%。这一结果强调了身体的日常节律在「少吃可助于长寿」效应中起着重要的作用 ! 图 1. 来源 Science 一、限制热量摄入延长寿命取决于进食时间以往研究发现,在保证营养摄入且无饥饿情况,通过降低

2022 年 5 月 12 日,曹彬和王健伟教授团队在《柳叶刀-呼吸病学》(The Lancet Respiratory Medicine)发表一项关于新冠住院患者出院 2 年随访结果研究,关注新冠肺炎长期影响。 该研究纳入 2020 年 1 月 7 日至 5 月 29 日期间武汉金银潭医院的 1192 名新冠住院患者,并分别在出院后 6 个月、1 年和 2 年进行随访。 研究结果表明:无论最初的疾病严重程度如何,患者的身体和心理健康都会随着时间的推移而改善。随访 2 年时,出院康复者中至少出现 1 种新冠肺炎长期影响的比例为 55%(650/1190),相较于 6 个月时明显下降(68%,777/1149),其中疲劳或肌肉无力最常见的症状。随访 6 个月时,存在呼吸困难(mMRC 评分≥1)比例为 26%(288/1104),到 2 年时这一比例明显降低至 14%(168/1191);存在焦虑或抑郁症状比例从 6 个月时 23%(256/110)降低至 2 年时的 12%(143/1191)。与普通人群相比,新冠康复者在初次感染 2 年后的健康状况往往较差,这表明一些患者需要更多时

