相关专题 引物 设计完成之后,需要用软件进行分析,找到最佳的引物对,采用Blast分析验证引物,可以知道序列的正确性,也能知道引物的特异性状况、引物的具体位置以及PCR产物的大小。 先找到需要设计引物的目标基因的在有引物序列的mrna 序列,可以通过全文(如最先发现该基因的文章),全文中有时可以找到大鼠c-jun mRNA序列的ACCESSION NUMBER,在PubMed中的“Nucleotide”中用

引物设计完成之后,需要用软件 进行分析,找到最佳的引物对,采用Blast分析验证引物,可以知道序列的正确性,也能知道引物的特异性状况、引物的具体位置以及PCR产物的大小。先找到需要设计引物的目标基因的在有引物序列的mRNA序列,可以通过全文(如最先发现该基因的文章),全文中有时可以找到大鼠c-jun mRNA序列的ACCESSION NUMBER,在PubMed中的“Nucleotide”中用此ACCESSION NUMBER就能找到序列。可以利用这个ACCESSION NUMBER
STS查找已经公布的引物序列(第二页) Part four: 如何运用BLAST进行序列比对、检验引物特异性(第三页) Part Five:5.1 从SNP的序列号入手找到其对应的基因序列和该SNP在基因组中的具体位置。5.2 查找目的基因的所有已公布SNP位点(第四页) 如果您感觉网页形式看起来太乱,请到 http://www.dxy.cn/bbs/post/view?bid=64&id=9681760&sty=1&tpg=2&age=0 下载PDF版本(此PDF版本为我实验

先找到需要设计引物的目标基因的在有引物序列的 mRNA 序列,可以通过全文(如最先发现该基因的文章),全文中有时可以找到大鼠 c-jun mRNA 序列的 ACCESSION NUMBER ,在 PubMed 中的“ Nucleotide ”中用此 ACCESSION NUMBER 就能找到序列。可以利用这个 ACCESSION NUMBER 在 PubMed 中的“ Nucleotide ”中搜索到序列后,点击右边的“ Links ”,再点击里面的“ Related
先找到需要设计引物的目标基因的在有引物序列的mRNA序列,可以通过全文(如最先发现该基因的文章),全文中有时可以找到大鼠c-jun mRNA序列的ACCESSION NUMBER,在PubMed中的“Nucleotide”中用此ACCESSION NUMBER就能找到序列。可以利用这个ACCESSION NUMBER在PubMed中的“Nucleotide”中搜索到序列后,点击右边的“Links”,再点击里面的“Related Sequences”就能找到所有的大鼠c-jun mRNA序列
相关专题 一步步教你设计简并引物 简并引物 设计都要注意些什么呢?用Primer5.0怎样设计兼并引物呢?下面是网上找到的详细的简并引物设计原则和Primer5.0 设计简并引物的方法可以参考下: 理论上说:由氨基酸 回导核酸序列,同时考虑到细菌使用密码子的偏爱性,以减少兼并度,但是这样得到的兼并引物的兼并密码子比较多,我建议你可以先从网上找到已经报道的比较近的几种菌的序列,然后比对,设计兼并引物,这样做没问题,我觉得
相关专题 一步步教你设计简并引物 简并引物设计 都要注意些什么呢?用Primer5.0怎样设计兼并引物呢?下面是网上找到的详细的简并引物设计原则和Primer5.0 设计简并引物的方法可以参考下: 理论上说:由氨基酸回导核酸序列,同时考虑到细菌使用密码子的偏爱性,以减少兼并度,但是这样得到的兼并引物的兼并密码子比较多,我建议你可以先从网上找到已经报道的比较近的几种菌的序列,然后比对,设计兼并引物,这样做没问题,我觉得还比较
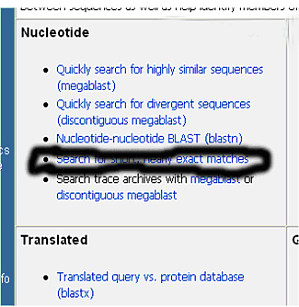
近段时间在“PCR技术讨论板”有不少关于RT-PCR引物设计的求助,所求助的引物有相当部分都是用于检测某一基因的表达的。其实,对于很多基因而言,用于检测其表达的RT-PCR引物已早有文献报道,我们完全可以利用这些已有的资源。我们所需要做的只是找到这些已发表的引物序列,通过Blast验证其是否正确、确定其特异性,用软件分析引物的好坏后直接使用就可以了。下面介绍2种搜索引物序列的方法,希望对大家有用。1. 通过搜索基因来找到所需引物序列 a. 打开“Entrez-PubMed”后,将“Search
测序都是从5'端进行的,正向和反向测序是指对DNA的两条互补链分别测序,通常两个方向测序结果经校读后完全一致才能认为得到可靠结果。生工测序结果一般都提供两个文档,一个是TEXT的序列文档,一个是用Chromas软件打开的ABI文档。 1.寻找引物 http://blast.ncbi.nlm.nih.gov/Blast.cgi 比对,去除引物序列,找到目的片段。 在DNAMan上进行比对,看引物能不能比对上(一个不变,一个反向互补),如果比不上,那可

。洗涤后离心时可两次分别将 EP 管置于不同的方向,便于彻底洗涤 RNA 样本。通常洗涤两次后可使测得的 OD 值较为理想。二、 利用「RNAstructure」软件引物,是进行荧光定量实验的必备条件。其特异性的问题可通过「BLAST」搞定,让人比较头痛的是引物可以形成「发夹结构」或「二聚体」(即在<75℃ 时出现溶解曲线的峰),影响扩增效率,最终导致实验结果不可用。那么,这个问题能否解决呢?有!设计引物的时候用「RNAstructure」预测一下引物的二级结构即可。那么,这个软件要如何使用

。洗涤后离心时可两次分别将 EP 管置于不同的方向,便于彻底洗涤 RNA 样本。通常洗涤两次后可使测得的 OD 值较为理想。二、 利用「RNAstructure」软件引物,是进行荧光定量实验的必备条件。其特异性的问题可通过「BLAST」搞定,让人比较头痛的是引物可以形成「发夹结构」或「二聚体」(即在<75℃ 时出现溶解曲线的峰),影响扩增效率,最终导致实验结果不可用。那么,这个问题能否解决呢?有!设计引物的时候用「RNAstructure」预测一下引物的二级结构即可。那么,这个软件要如何使用
Oligo 6 作为目前最好、最为专业的引物设计软件,Oligo 的功能很强大,他主要功能有:普通引物对的搜索、测序引物的设计、杂交探针的设计以及评估「引物对」质量的功能。 如何使用Oligo6 请参看:「两分钟教你学会 Oligo 7」 BLAST BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或 DNA 数据库中进行相似性比较的分析工具。BLAST 程序能迅速与公开数据库进行相似性序列比较。
:① 模板的选择。如果以DNA为模板设计引物, 首先在Genebank中找到与待 分离新基因同源的其它物种的该基因。利用http://www.ncbi.nlm.nih.gov/blast/ 的Blast工具和http://www.ebi.ac.uk/elustalw/ 的Clustalw工具把已检索到的基因进行同源性比较,根据比 较基因组 定位的原理,选择研究深入、标记稠密的人和哺乳动物(如小鼠)保守功能基因DNA序列设计引物[51,该引物区段要求在各物种间绝对保守,差异不要大于2 bp
的一系列引物分别在Genebank中进行回检。也就是把每条引物在比对工具(http://www.NCBI.nlm.nih.gov/Blast/) 的blastn中进行同源性检索,弃掉与基因组其它部分同源性比较高的引物,也就是有可能形成错配的引物。一般连续10 bp以上的同源有可能形成比较稳定的错配,特别是引物的3’端应避免连续5-6 bp的同源。 二是以mRNA为模板设计引物时要先利用生物信息学的知识大致判断外显子与内含子的剪接位点(例如http://CCR-081
将两个蛋白放在一起直观地进行三维结构上的比较,如下图中是将两种核酸外切酶的三维结构通过VAST对准得到的结构比较图:同样,Cn3D在结构比较方面也能利用其内嵌的Blast搜索引擎直接访问Genbank数据库找到具有局部相似性的结构数据,并在三维结构图中显示出二者具有相似性的结构区域 12. Seqverter 1.3 Seqverter 1.3使用简单,填写输入文件和输出文件名就可以完成格式转换。而且能够转换的格式非常之多是其它软件所无法比拟的。另外,它可在线升级以读写更多
,因此可以提髙扩增的特异性。以胸腺嘧啶结尾的引物的特异性有降低的趋势。 高 GC 含量片段的扩增 为了扩增高 GC 含量的 DNA,应该设计 Tm 值(最好为 75~80℃) 较高的引物。髙 GC 含量的 DNA 双链完全解开所需的温度明显较一般的 DNA 高。温度较低时,PCR 复制子的两条单链有较快地重新退火的趋势,可与引物退火相互竞争。因此,PCR 过程中退火温度较髙有助于引物退火,因此可以提髙扩增效率。 非目标同源 应利用 BLAST(http: //www
相关专题 引物设计 一步步教你设计简并引物 近日看到两位虫友发帖交流有关CODEHOP方法设计简并引物,这让我想起两年前因为需要获得几个家族的未知序列特异基因而学习利用codehop方法设计简并引物的经历。当时,通过常规方法设计出来的简并引物扩增效率极低,而且特异性非常差,为此纠结了好一段时间。后来从一同事那里得知设计简并引物可以利用codehop方法,但是他对该方法也只是听说没有用过,所以我只能自己在网上查找资料并且试图询问有经验
与这些DNA片段进行杂交,在含有特异性模板的区域就会出现杂交带,可以初步鉴定基因组中是否含有目的基因。我们也可以将酶切后的基因组DNA片段全部克隆入适当的载体中,制成基因组文库,再用特异性探针与基因组文库中的不同克隆进行杂交,阳性克隆即表示克隆到的DNA片段含有特异基因序列。将阳性克隆测序,用第一个克隆片段的末端分离下一个克隆片段,然后利用DNA片段间的重叠顺序来鉴定其他克隆。这样一步步走下去,最终可得全部基因序列,这种技术叫做染色体步移(chromosome walking)。 (二)采用
行观察,比其他软件要简便。而Cn3D主要的特点是能够将两个蛋白放在一起直观地进行三维结构上的比较,如下图中是将两种核酸外切酶的三维结构通过VAST对准得到的结构比较图:同样,Cn3D在结构比较方面也能利用其内嵌的Blast搜索引擎直接访问Genbank数据库找到具有局部相似性的结构数据,并在三维结构图中显示出二者具有相似性的结构区域 12. Seqverter 1.3 Seqverter 1.3使用简单,填写输入文件和输出文件名就可以完成格式转换。而且能够转换的格式非常
相关专题 引物设计 RT-PCR引物 设计的主要的过程包括如下:首先查找目的基因的序列并编辑,其次是利用软件进行引物设计并选择合适的上下游引物序列。详情如下: 在NCBI上搜索到该基因,找到该基因的mrna ,在CDS选项中,找到编码区所在位置,在下面的origin中,Copy该编码序列作为软件查询序列的候选对象。 打开Primer Premier5,点击File-New-DNA sequence, 出现
关于丁香通
公司信息
个人用户
企业机构


