想要 PCR 不踩「坑」,学会这 3 招就对了...
丁香园
PCR 是一个坑,坑里埋了好多研究僧……
PCR 是众位「研究僧」日常实验中最常用的技术,可谓是「研究盛宴」中的一道「小菜」。可也有不少童鞋反映,这道菜不好吃,一不小心就「小河里翻船」。
那么,我们该如何把这道菜「吃好」、跳出这个「坑」呢?
诸位看官莫急,待我送君「三大法宝」。
一、确保 RNA 样本合格
常见的 RNA 提取方法有 TRIzol 法、吸附法,这两个方法各有所长,但都不能保证提取的 RNA 样本一定合格。
因而,提取完成后应测定 RNA 的浓度及吸光度。通常,在 260,280,230 nm 处分别测定其吸光度(A260、A280、A230)并计算其比值(A260 /A280,A260 /A230)。

那么,这几个数值及其比值分别有何意义?
A260 为核酸的吸光度,A280 为蛋白质的吸光度,A230 为其他杂质(多糖等)的吸光度。纯 DNA 的 A260 /A280 为 1.8,纯 RNA 的 A260 /A280 为 2.0。
如果 OD 比值不在范围内该如何解决呢?
再次洗涤样本!
75% 乙醇沉淀 RNA 后重新吸附、洗涤;若采用 TRIzol 法提取 RNA,可直接洗涤两次。洗涤后离心时可两次分别将 EP 管置于不同的方向,便于彻底洗涤 RNA 样本。通常洗涤两次后可使测得的 OD 值较为理想。
二、 利用「RNAstructure」软件
引物,是进行荧光定量实验的必备条件。其特异性的问题可通过「BLAST」搞定,让人比较头痛的是引物可以形成「发夹结构」或「二聚体」(即在<75℃ 时出现溶解曲线的峰),影响扩增效率,最终导致实验结果不可用。
那么,这个问题能否解决呢?
有!设计引物的时候用「RNAstructure」预测一下引物的二级结构即可。

那么,这个软件要如何使用呢?
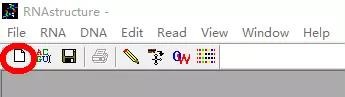
1. 打开「RNAstructure」,单击左上角的「new sequence」按钮:
2. 新建序列文件,会出现一个对话框,输入待检测的引物序列:

3. 单击「Fold as DNA」,「save changes」,出现新的对话框后直接点击「Start」:
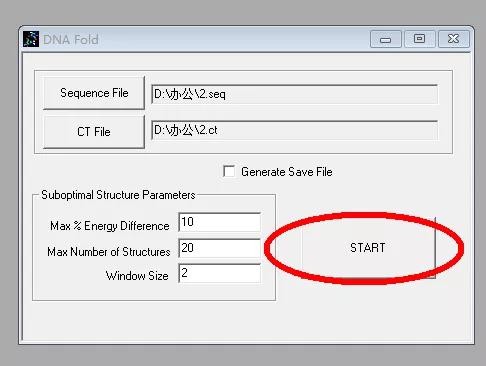
4. 无需更改其他参数,再次点击「Draw structure」,即可得到目标序列的二级结构:
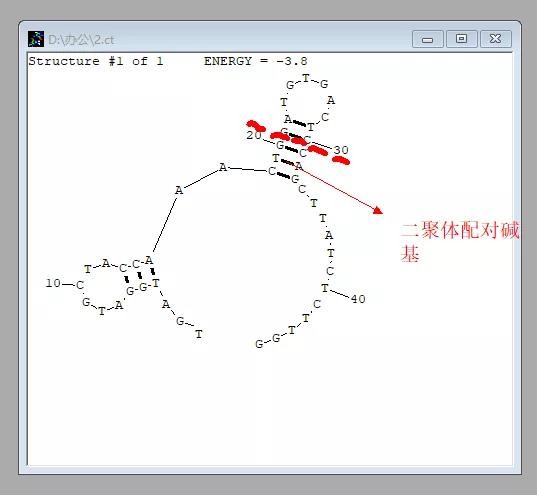
若考察引物能否形成发夹结构,则直接检测 Forward 或 Reverse 引物即可。
若考察引物能否形成二聚体,则把两条引物一起输入,视作一条序列进行检测即可。
在得到二级结构图后,只要没有连续 4 个以上的配对即可;若一条引物长度为 20 bp,在 18~22 处有连续 5 个的碱基配对,则可「手动拆解配对」,即保证引物为两条链时的配对不多于 4 个即可。
三、保证扩增效率符合要求
有童鞋反映,两个同类基因测试同一批样本,理论上趋势应该一致,结果却是两种趋势,应该相信哪个呢?
不知道各位看官有没有遇到类似的问题?这类问题该如何解决呢?
先说说出现这类问题的原因吧:
扩增条件非最佳;
样本中含有抑制扩增的物质,如乙醇。
一对引物有其特定的 Tm 值,但 Tm 值时未必是其最佳扩增温度。温度过高时,扩增效率过低,差异部分的目的基因不能被扩增,导致「假阴性」结果的出现。
那么,如何确定扩增效率是否满足要求呢?
通常,拿到新引物后应从 Tm- 5℃ 寻找其适宜的扩增温度,在某温度起为特异性扩增。
然后,在特异性扩增的温度下检测扩增效率:将 cDNA 梯度稀释(一般为 2×),测试稀释后的样本 Ct 值差异是否为 1 左右。
若差值为 1,则扩增效率良好,可进行正式实验。
若在特异性扩增的温度范围内不能使 Ct 值差异为 1,则需考虑重新设计引物。