简介
插入缺失率可以用 SURVEYOR 核峻酶测定法或新一代测序技术测定,新一代测序技术更适合对大量 sgRNA 作用杷标位置进行采样。
材料与仪器
器材:Illumina MiSeq 等
试剂:
① 用于扩增二代测序的文库的引物,用于扩增二代测序插入缺失的引物。长 度大于 60 bp 的引物可以订购为 4 nmol 超级单体(Integrated DNA technologies)
② KAPA HiFi HotStart Ready Mix, 2X(Kapa Biosystems, cat. no. KK2602)。 关键:为了最小化扩増 Oligo 时的错误,使用高保真聚合酶非常重要。其他 高保真聚合酶,例如 PfuUltra U(Agilent)或 Kapa HiFi(Kapa Biosystems), 也可以用作替代品。为了扩增用于插入缺失分析的 gDNA, 建议使用 KAPA HiFi HotStart ReadyMiX。
③ Qubit dsDNA HS 检测试剂盒(Thermo Fisher, cat. no. Q32851)
④ NextSeq 500/550 High Output Kit v2(150 cycle; Illumina, cat. no. FC-404- 2002)
⑤ MiSeq Reagent Kit v3(150 cycle; Illumina, cat. no. MS-102-3001)
⑥ MiSeq Reagent Kit v2(300 cycle; Illumina, cat. no. MS-102-2002)
⑦ PhiX Control Kit v3(Illumina, cat. no. FC-110-3001)
⑧ 氢氧化钠溶液,10 mol/L(Sigma-Aldrich, cat. no. 72068-100 mL)。注意: 穿防护服并避免接触 10 mol/L 以氧化钠,如果皮肤接触,眼睛接触,食入和 吸入会非常危险
⑨ Tris, pH 7.0(Thermo Fisher, cat. no. AM9850 G)
步骤
二代测序确定 sgRNA 文库的 sgRNA 分布的基本过程可分为如下几步:
A. 文库 PCR 用于二代测序。可使用 Illumina 衔接子序列扩增 sgRNA 靶区域。要为二代测序准备 sgRNA 文库,需为 10 个二代测序 -Lib-Fwd 引物和 1 个二代测序 -Lib-KO-Rev 或二代测序 -Lib-SAM-Rev 条码引物分别设置一个反应,如表 9.13 所示。
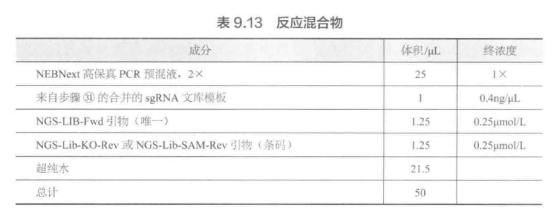
关键步骤:为每个文库使用不同的下游引物和独一无二的条形码,可以 在一次 NextSeq 或 HiSeq 运行中聚合和测序不同的文库。与在多重 Miseq 中运行相同数量的库相比,这更高效且更划算。
关键步骤:为了使扩增 sgRNA 的错误最小化,使用高保真聚合酶非常重要。 其他高保真聚合酶,例如 PfiiUltra H(Agilent)或 Kapa HiFi(Kapa Biosystems), 也可以用作替代品。
B. 使用表 9.14 所示循环条件进行 PCR。
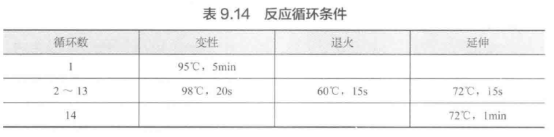
C. 反应完成后,根据生产厂家的说明,使用 QlAquick PCR 纯化试剂盒, 将 PCR 反应进行混合,对 PCR 产物进行纯化。
D. 定量 PCR 产物,并在 20 g/L 琼脂糖凝胶上电泳 2 μg 产物。成功敲除文 库应产生约 260?270bp 的产物,对于激活文库应产生约 270~280bp 的产物。 使用 QIAquick 凝胶回收试剂盒回收凝胶。凝胶提取的样品可以在 -20 ℃ 保存数月。
E .使用 Qubit dsDNA HS 分析试剂盒对凝胶提取样品进行定量。
F. 根据 Illumina 用户手册,对 Illumina MiSeq 或 NextSeq 上的样品进行 80 个 read 1(forward)循环和 8 个 index 1 循环的测序。推荐在 MiSeq 上使用 5% PhiX 对照或在 NextSeq ± 使用 20% PhiX 进行测序,以改善文库多样性,并争 取覆盖文库中每个 sgRNA 大于 100 个读长。
G. 使用 count_spacers.py 分析测序数据。安装 python 2.7(https://www. python.o 回 downloads/)禾口 biopython(http://biopython.Org/dist/dcM:s/instaIl/install.html) 准备一个包含引导间隔序列的 csv 文件,每行对应一个序列。
H. 要确定间隔分布,请使用表 9.15 所示可选参数运行 python
 count_spacers.pyocount_spacers 运行后,间隔子读取结果将写入输出的 csv 文件。相关的统 计信息包括完全向导匹配的数量、非完全向导匹配的数量、无键的测序读取 数量、处理的读取数量、完全匹配的向导百分比、未检测到的向导的百分比 和倾斜比,将被写入 statistics.txto 理想的 sgRNA 文库应具有超过 70% 的完 全匹配的引物,少于 0.5% 的未检测到的引物以及倾斜比小于 10。关键步骤: 人类 SAM 文库在引导间隔区序列之前没有鸟瞟吟,因此在分析这些库时,请确保使用参数 -no-g 运行脚本。
count_spacers.pyocount_spacers 运行后,间隔子读取结果将写入输出的 csv 文件。相关的统 计信息包括完全向导匹配的数量、非完全向导匹配的数量、无键的测序读取 数量、处理的读取数量、完全匹配的向导百分比、未检测到的向导的百分比 和倾斜比,将被写入 statistics.txto 理想的 sgRNA 文库应具有超过 70% 的完 全匹配的引物,少于 0.5% 的未检测到的引物以及倾斜比小于 10。关键步骤: 人类 SAM 文库在引导间隔区序列之前没有鸟瞟吟,因此在分析这些库时,请确保使用参数 -no-g 运行脚本。
来源:丁香实验