





3 个回答

sswei
有帮助
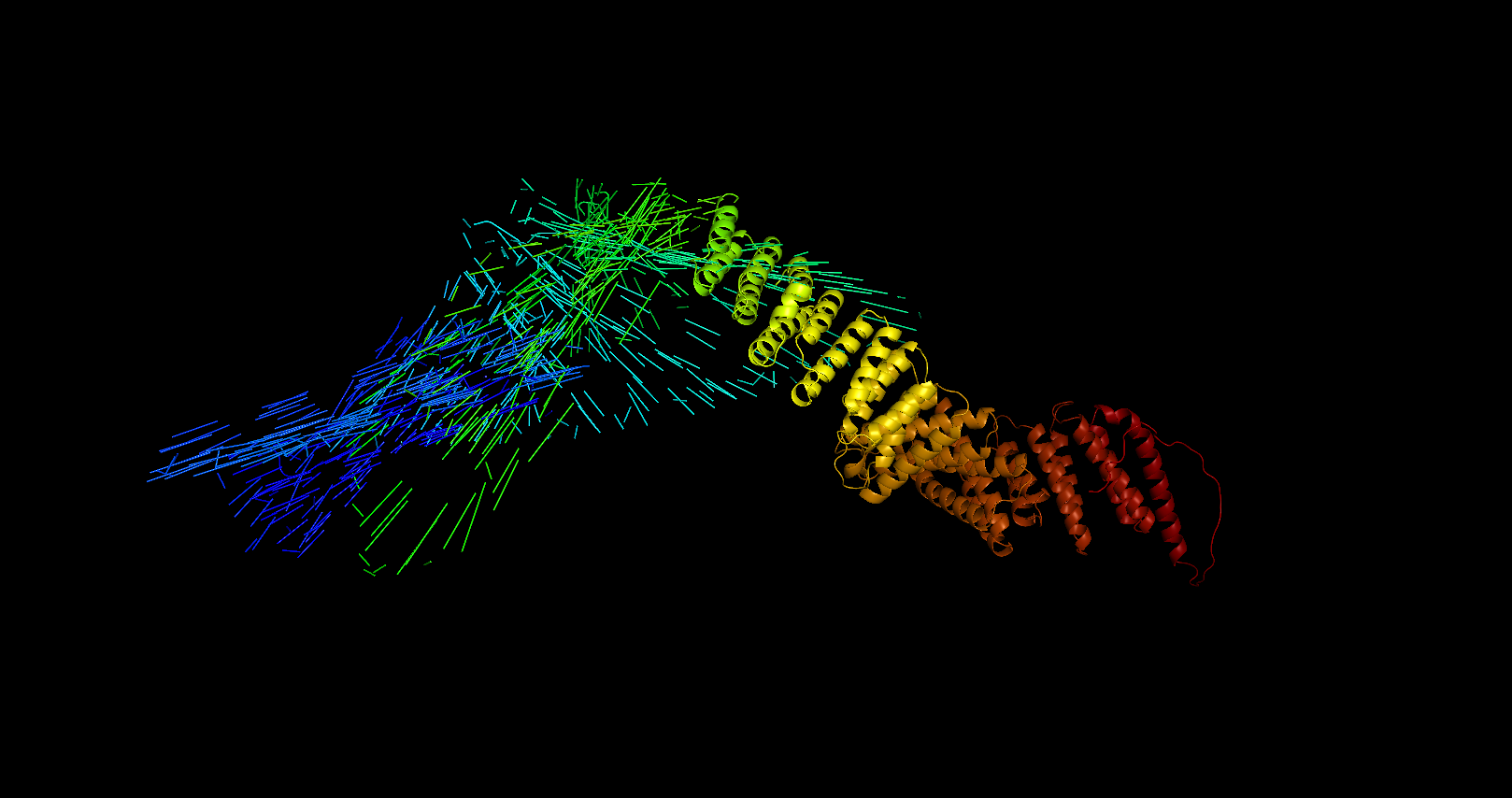
ZDOCK生成的结果文件可能存在一些错误或者不完整的信息,例如缺失原子坐标、残基信息不完整等。这可能会导致Pymol无法正确显示结果。解决方法是检查结果文件是否存在问题,并尝试重新生成结果文件或者使用其他对接软件进行分析。显示参数设置问题。Pymol的图像显示可以通过设置不同的参数进行调整,例如颜色、透明度、视角等。如果显示参数设置不当,可能会导致图像不太正常。解决方法是检查显示参数设置是否合适,并进行适当的调整。”

土井挞克树
有帮助
考虑是通道没有选择正确,可以选择单向通道对接

loveliufudan
有帮助
可能有几个原因导致:
1. 数据格式问题:确保你的对接结果数据文件(如PDB文件)与PyMOL兼容。检查文件格式和结构是否正确,并确认文件中包含所需的原子坐标和信息。
2. 数据处理问题:在将对接结果导入PyMOL之前,可能需要对数据进行一些预处理。例如,可以根据能量或分数进行排序或筛选,只保留最优的对接构象或复合物。这样可以减少杂乱性并更好地展示重要的相互作用。
3. 选择显示的结构或链:对接结果通常包含多个构象或复合物,可能涉及多个蛋白链或配体。确保正确选择要在PyMOL中显示的结构或链,并根据需要进行标记或分离。
4. 可视化参数设置:在PyMOL中,你可以调整可视化参数来优化显示效果。尝试调整可视化参数,例如球和棍的大小、颜色、透明度,或者调整背景和光照设置,以获得更清晰和易读的图像。
5. 数据解释问题:如果对接结果显示为杂乱的结构,可能是由于蛋白质间的高度柔性或多样性,或者是由于对接算法的特性导致的。在这种情况下,你可能需要更仔细地分析结果,关注重要的相互作用、结合位点和构象变化。




相关问答
提问
扫一扫

实验小助手

扫码领资料
反馈
TOP
打开小程序