使用 pymol 对蛋白结构进行 align
ZeroDesigner
Align
align 首先执行序列比对,然后进行结构叠加,进行多次迭代以便进行微调,在蛋白序列相似性大于 30% 的时候可以达到良好的效果。
用途
Align 常常在结构生物学以及虚拟筛选中使用,当对不同的蛋白结构并对其进行比较时,我们就可以使用 align 比较蛋白结构,查看两者之间的差异,这个结构上的差异有一个量化的指标就是 RMSD。它的概念和计算方式,都会在下面列出。目前,pymol 是一个很流行的三维蛋白结构显示工具。本次的目的是,使用 pymol 对蛋白结构进行 align,结果可以通过肉眼观测或者 RMSD 进行量化。
用法
align mobile, target [, cutoff [, cycles
[, gap [, extend [, max_gap [, object
[, matrix [, mobile_state [, target_state
[, quiet [, max_skip [, transform [, reset ]]]]]]]]]]]]]
解释:
mobile = 字符串:需要移动的对象名
target = 字符串:目标的对象名
cutoff = 浮点数:截断值,默认 2.0
cycles = 整数:最大循环数,默认 5
gap, extend, max_gap: 序列对比参数
object = 字符串:创建的一个比较对象名,默认无
matrix = 字符串:序列比对的替换矩阵的文件名,默认 BLOSUM62
mobile_state = 整数:移动选择的对象状态,默认全状态
target_state = 整数:目标选择的对象状态, 默认全状态
transform = 0/1: 是否做叠加,默认 1
Alignment Objects
可以使用 object = somename 参数创建 align 对象。
align 对象可以:
(1)对比序列查看器
(2)对比原子对结果展示 3D 查看器中的线条
(2)可以保存到 clustalw 序列比对文件
RMSD
单位是埃
RMSD,root-mean-square deviation,也就是均方根偏差。
原子位置的均方根偏差是叠加蛋白质的原子(通常是骨架原子)之间的平均距离的量度。注意,RMSD 计算可以应用于其他非蛋白质分子,如小的有机分子。在球状蛋白质构象的研究中,通常在刚体进行完叠加后通过计算 Cα 原子坐标之间的 RMSD 来表征三维结构的相似性。
等式:
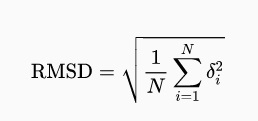
其中 δi 是原子 i 与参考原子之间的距离。计算时通常只计算为骨架重原子 C,N,O 和 Cα 或有时仅计算 Cα 原子。
例如给出两套原子坐标,v 和 w,计算他们的 RMSD 就是,如下。

通常,RMSD 用作两种或更多种蛋白质结构之间相似性的定量测量,通常越低越好。
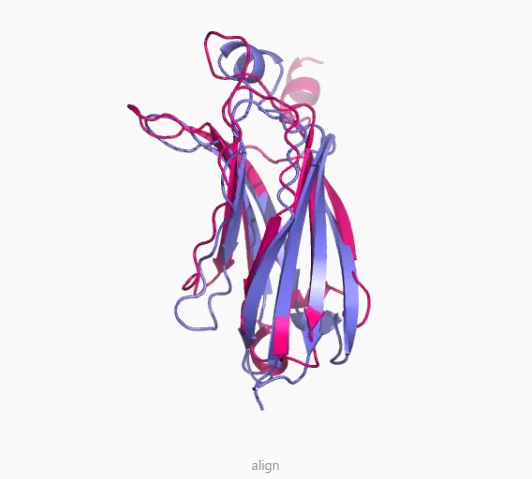
Examples
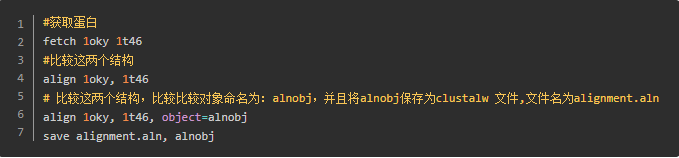
结果
alignment.aln 文本内容

alignment
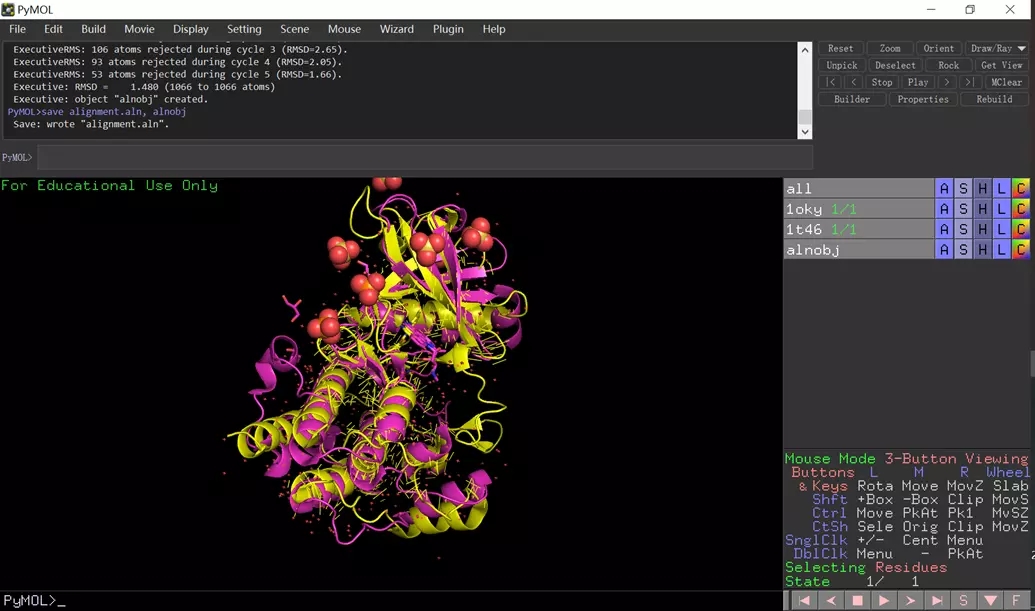
pymol 显示结果
PS:附加鼠标操作流程
1t46 右侧的 A 按钮 -->align-->to molecule-->1oky


![4-[2-(2-Amino-4,7-dihydro-4-oxo-1H-pymol[2,3-d]pyrimodin-5-yl)ethyl]benzoic acid methyl ester厂家](https://img1.dxycdn.com/2019/0710/340/3355961533879910862-14.gif!wh200)

![4-[2-(2-Amino-4,7-dihydro-4-oxo-1H-pymol[2,3-d]pyrimodin-5-yl)ethyl]benzoic acid methyl ester155405-80-4](https://img1.dxycdn.com/2019/0702/582/3354462285350744046-14.gif!wh200)
![4-[2-(2-Amino-4,7-dihydro-4-oxo-1H-pymol[2,3-d]pyrimodin-5-yl)ethyl]benzoic acid methyl ester155405-80-4](https://img1.dxycdn.com/2019/0626/467/3353331538573582084-14.gif!wh200)