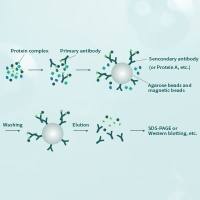
免疫沉淀-蛋白印迹法
丁香园论坛
Methods
2.1. Material and reagents
Duolbecco’s phosphate buffered saline (PBS), Medium 199 with Earle’s salts and 25mM HEPES, collagenase Type II (from Clostridum histolyticum), antibiotic/antimyotic solutuion (containing 10,000U penicillin, 10,000µg streptomycin, 25µg amphotericin B per ml in saline) and foetal bovine serum from Invitrogen Life Technologies (Paisley, UK).
Endothelial Cell Growth Supplement (ECGS) from bovine neural tissue was kindly provided by Mrs.S.Joy (Centre for Cardiovascular Biology & Medicine, King College London, UK).
Rainbow recombinant protein molecular protein markers, ECL western blotting detection reagents were purchased from Amersham Pharmacia Boutech (Little Chalfont, Buckinghamshire, UK).
Protogel 30%acrylamide solution, 10x stock Tris/ Glycine SDS buffer were from National Diagnostics, (Hull, UK).
eNOS (NOS 3) affinity purified rabbit polyclonal antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, Calafornia, USA).
Goat Anti-Rabbit horseradish peroxidae (HRP)-conjugated IgG; Rabbit Anti-Mouse horseradish peroxidase were from Perbio Science UK Ltd (Cheshire, UK).
Heparin (sodium salt Grade A), glycerol, methanol, Bromophenol Blue (sultone form), 2-mercaptoethanol, glycine, leupeptin, aprotenin, NaCl, Tris, TWEEN-20, Folin Ciocaltew Reagent, sodium dodecyl sulphate (SDS), NaTartrate, NaOH, NaCO3, bovine serum albumin, HCl, protein sepharose A, butanol, CuSO4,, 10x stock Trypsin/EDTA solution, tetramethylethylenediamine (TEMED), ammonium persulphate (APS), photographic film, photographic film developer, photographic film fixer, gelatin 2% solution, Hsp-90 affinity purified mouse monoclonal antibody, general actin mouse monoclonal antibody, ß-actin mouse monoclonal antibody, caveolin mouse monoclonal antibody were obtained from Sigma Aldrich Co. Ltd (Gillingham, Dorset, UK).
2.2. Human Umbilical Vein Endothelial Cell (HUVEC) Isolation and Culture
Following cannulation of the umbilical vein, 50ml of warm sterile phosphate buffered saline (PBS) was flushed through the vein using a 0.2mm filter in order to remove all blood present in the vein. To ensure the vein was empty, the umbilical cord was gently massaged. Following this, both ends of the cord were clamped, and the vein filled with warm sterile 1mg/ml collagenase type II (activity: 1.5 units/mg solid) solution, using the same cannula and another 0.2mm filter. After a further 10 mins. The ‘digest’ was then collected into a sterile 50ml centrifuge tube. More warm sterile PBS was passed through the vein and collected in the same centrifuge tube, to ensure maximal retrieval of endothelial cells.
The suspension was centrifuged for 5 mins. at 600G at room temperature. This resulted in the formation of a pellet, containing HUVEC as well as other debris. The supernatant was aspirated, and the pellet resuspended in 5ml of warm sterile growth medium. This growth medium consisted of Medium 199(with 5% HEPES, Earle’s Salts, L-glutamine, L-glutamine tryptophan and phenol red) containing added Foetal Calf serum (20%), Endothelial Cell Growth Supplement from bovine neural tissue (120mg/ml), Heparin sodium Grade A (0.01%), penicillin (500i.u./ml), streptomycin (500mg/ml) and amphotericin B (1.25mg/ml).
The growth medium and cell suspension was gently mixed and transferred into a gelatin coated sterile culture flask with a 25cm2 surface area (T25). This was left overnight at 37°C in a sterile atmosphere of 95%air and 5% CO2, to allow the attatchment of endothelial cells to the flask. The following day, the medium was removed and the cells were washed with warm sterile PBS in order to remove any unwanted cell types and debris. This was followed by adding 5ml of fresh warm sterile growth medium.
2.2.2. Feeding of cells
The cells were fed approximately every 48 hours, by replacing the growth medium with new warm sterile growth medium. When not being fed the cells were incubated at 37°C in a sterile atmosphere of 95% air and 5% CO2. This continued for several days until the cells formed a confluent monolayer (Fig.1.), at which point they were ready for passaging.
2.2.3. Passaging of cells
In order to passage HUVEC, the growth medium was aspirated and the cells washed once with warm sterile PBS, followed by another wash with warm sterile Trypsin/EDTA solution (1x solution). The Trypsin/EDTA solution was immediately removed, a small amount (approx 0.5ml) of the solution was left in the flask. The flask was then incubated at 37°C for a few minutes, allowing the Trypsin/ EDTA solution to cause the cells to detach from the flask surface. The cells were carefully observed under a microscope to check that detachment had occured. The cells were then resuspended in 5ml of warm sterile growth medium, added to the Trypsin/EDTA solution. This cell suspension was then transferred equally into sterile culture flasks. Extra growth medium was added so there was approximately 5ml in a T25 flask, 15ml in a T75 flask or 25ml in a T125 flask. The cells were fed as described earlier until confluent. Only confluent cells that had not been passaged more than two times were used in the experiments outlined here.
2.2.4. Harvesting and lysis of cells
All growth medium was removed from the culture flasks, and the cells washed in cold sterile PBS twice. The culture flasks were then placed in a sonicator for 5 minutes, causing the cells to detach from the flasks and to lyse. 130ml of lysis buffer was added to each culture flask. The lysis buffer consisted of Tris(25mM), NaCl (50mM), Aprotenin(1mg/l), Leupeptin (10mg/ml) and PMSF (1mM). The flasks were then placed on an ice tray for five minutes, after which all the cells were scraped off the flasks using a rubber policeman. The cell suspension was transferred into Eppendorf tubes, which were then heated at 100°C for 5 minutes.
2.2.6. Determination of protein concentration
Twelve standard concentrations of bovine serum albumin (BSA) ranging from 0.1mg/ml to 4mg/ml were made up in Eppendorfs. Using a 96-well plate, 25ml of each standard and of each unknown sample were pipetted into separate wells. Then 75ml of a solution of Lowry reagent A plus Lowry reagent B, mixed in a ratio of 100:1, was added to each well. (Lowry sol A consists of 2% NaCO3, 0.4% NaOH, 0.16% Na tartrate and 1% SDS. Lowry sol.B is a 4%CuSO4 solution.) The well plate was left at room temperature for 20 minutes on a shaking stirrer.
Then 7.5ml of Folin Ciocaltew Reagent was added to each well. The well plate was then left at room temperature for a further 30 minutes. Absorbencies were read at 750nm (A750) in a plate-reading spectrophotometer.
A protein standard curve was constructed by linear regression of the A750 readings of the standards; using this standard curve and the A750 of each sample, the corresponding protein concentration could be determined. All data was analysed using Microsoft EXCEL 97.
2.3 Immunoprecipitation of endothelial cell lysates
Endothelial cell lysates, containing approximately 300mg of protein as measured above, were incubated with the relevant antibody (5ml for eNOS antibody, 25ml for general actin antibody) overnight at 4°C. 50ml of hydrated Protein sepharose A beads were then added, and incubation continued for a further 2 hours at 4°C. [The Protein Sepharose A beads had previously been hydrated by leaving a solution of 100mg protein sepharose A and 15ml TBS (22.3g Tris, 8g NaCl, 1M HCl) on a shaking stirrer overnight at 4°C, and then centrifuging the solution at 2000G, for 15 mins., at 4°C and washing the pellet with TBS three times.]
This mixture was centrifuged at 600G, the supernatant was removed and the pellet washed with PBS containing 0.1% Tween 20 solution (PBS-T). This was repeated 6 times and the final pellet was stored at -20°C.
2.4 SDS-Polyacrylamide Gel Electrophoresis
2.4.1 Sample preparation
Each sample consisting either of endothelial lysate (10mg protein) or immunoprecipitate (prepared as above) was mixed with 30ml of SDS-PAGE sample buffer (72.5% water, 12.5% 0.5M Tris pH 6.8, 10% Glycerol, 0.02g/ml SDS, 0.001g/ml Bromophenol Blue, 5% Mercaptoethanol). This mixture was then heated at 100°C for 4 minutes and vortexed. This was done in order to denature the protein sample.
2.4.2 Electrophoresis gel preparation
Bio-Rad mini gel equipment was used. The glass plates were cleaned with 70% ethanol. 1.5mm deep spacers were used. After setting up all the apparatus a freshly made 7.5% or 13% acrylamide separating gel (composition detailed in Fig.8) was added into the glass sandwich and allowed to set. A mixture of 50% butanol was poured on top of the separating gel while it set. Once the gel had set, this water saturated butanol mixture was discarded, and a 3% acrylamide stacking gel (composition detailed in Fig. 8) was poured on top. A 1.5mm deep comb was inserted into the stacking gel, which was then left to set.
7.5%Acrylamide gel 3%Acrylamide gel 13% Acrylamide gel
Protogel(30% Acrylamide) 5.0ml 1.0ml 8ml
Tris (1M) pH 8.8 7.5ml 1.4ml 4.5ml
Distilled water 7.5ml 7.8ml 7.5ml
20%SDS solution 5.0ml 50µl 0.1ml
10% APS solution 67µl 50µl 67µl
TEMED 13µl 20µl 13µl
After the stacking gel had set, the combs were removed and the wells formed were washed out with Tank Buffer (purchased as a 10x stock of Tris/Glycine/SDS buffer from National Diagnostics). The gel was then placed into the electrophoresis apparatus, which was then filled with tank buffer.
Each well was loaded with an appropriate amount of sample. One well was loaded with 10ml of full range rainbow marker in all experiments. This is used as a molecular weight standard and has a molecular weight range of 10-250 kDa. All other empty wells were loaded with 30ml of SDS-PAGE sample buffer.
The electrophoresis equipment was connected to a electricity supply of 100V and the gel was left to run until the bromophenol blue dye front reached the bottom of the gel.
2.5 Western blotting
Following SDS-PAGE, the gel was removed from the apparatus and the stacking gel discarded. One PVDF membrane and two pieces of filter paper were cut to the dimensions of the remaining gel. The PVDF membrane was placed in methanol for 1 minute and then washed in distilled water several times. The pieces of filter paper, gel and PVDF membrane were placed in transfer buffer (3.9mM Glycine, 4.8mM Tris, 0.00375% SDS, 2% methanol) for 5-10 minutes.
All these items were then layered on a transfer machine so that the bottom layer was filter paper, followed by the membrane, then the gel and lastly another filter paper. A roller was used to ensure that all bubbles were expelled. The transfer machine was connected to an electricity supply for 3 hours. The current needed (in mA) was calculated by multiplying the area of the gel (in cm2) by 0.8. In this way, proteins were electroblotted, resulting in complete transfer from the gel onto the PVDF membrane, as illustrated in Fig 2.