【操作步骤】6.2 分文章作者,亲手教你如何轻松搞定生信文章
科研论文时间
大家好,今天跟大家分享一篇文章「DDX60 is associated with glioma malignancy and serve as a potential immunotherapy biomarker」。
该文章是由笔者于 2021 年 2 月发表于《Frontiers in Oncology》杂志上的,目前该杂志中科院分区 2 区,影响因子 6.2 分。
今天就通过该文章跟大家分享一下肿瘤方向生信文章的套路及绘图方法~
信息发展到这个年代,许多科研道友仍然闻「代码」色变,总想走不用代码发文章的捷径。
殊不知,其实代码才是捷径,许多软件复杂的操作只需一行代码就能轻松搞定。
闲话少说,上干货~
总体思路
首先我们来看一下文章标题「DDX60 is associated with glioma malignancy and serve as a potential immunotherapy biomarker」,就是说 DDX60 这个基因在胶质瘤中很重要,能够提示胶质瘤患者预后不良,且与胶质瘤免疫应答相关。
听起来有点玄妙,那么具体是怎么操作的呢?
先通过 Results 的小标题来看一下总体套路:
1、目标基因在胶质瘤中表达增高
2、目标基因高表达能提示胶质瘤患者预后不良
3、目标基因与多种生物过程密切相关
4、目标基因作用于炎症反应和免疫应答
5、目标基因与胶质瘤患者的免疫检查位点关系密切
通过这一系列小标题,发现文章套路了吗?
总结而言,扩展到各种肿瘤的生信研究,大致思路就是:某基因的表达情况—生存预后分析—基因通路富集—富集结果验证—结合临床应用。
总体思路有了把握,那么具体如何执行呢?
Figure 1
首先,该文章通过「http://ualcan.path.uab.edu/」网站查找了目标基因在不同癌症中的差异表达情况,证明了该基因在多种癌症中的表达量较正常组均有所升高。
网站原始下载图片如下:
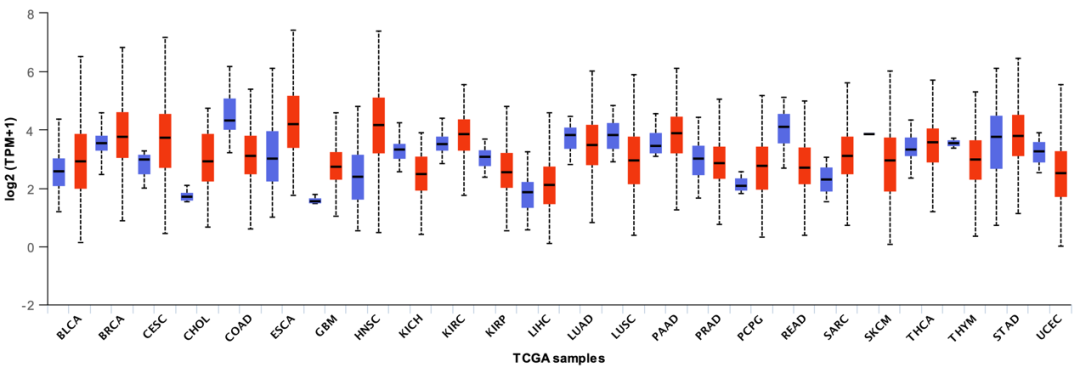
图片来源:文献截图
其次,该文章下载了 TCGA 的原始数据进行验证,并利用 R 语言进行绘图。
研究胶质瘤,分组情况一般是传统的病理级别(胶质瘤可分为 I-IV 级)、2016 版 WHO 分组情况(经典型、间质型、神经元型、前神经元型),以及临床常用的分子标志物表达情况(MGMT 启动子甲基化、ATRX 表达情况、TERT 表达情况、IDH1 表达情况)。
当然,2021 年 WHO 又更新了脑肿瘤的分类,那是后话,不体现在本文中。
组间差异表达的形式可以有多种图形及其变种,比如传统的箱式图,如空心箱式图、实心箱式图,还可以用箱式图结合散点图,如本文 Figure 1 B、Figure 1 C。
此外,小提琴图(Violin 图)也是近年来文献中常见的图形,其特点是更加直观地展示了数据的分布情况,如本文 Figure 1 D-G。绘图时建议多尝试几种,选择效果比较漂亮的放到你的 Paper 中。
由于篇幅原因,本文 R 语言绘图代码仅展示主要部分:
箱式图:
ggplot (grade_TCGA, aes (x = Grade, y = DDX60, fill = Grade)) +
stat_boxplot (geom = 'errorbar', width = 0.08) +
geom_boxplot (outlier.fill = "black", outlier.shape = 2, outlier.size = 0.1)
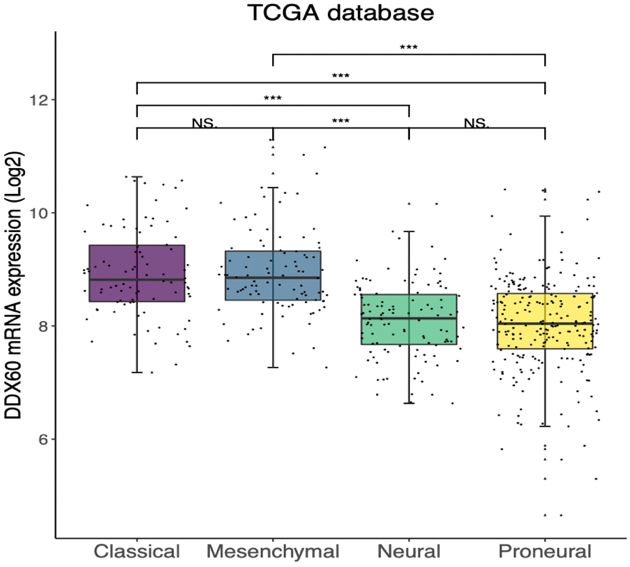
图片来源:文献截图
小提琴图:
ggplot (grade_TCGA, aes (x = Histology, y = DDX60, fill = Histology)) +
geom_violin (trim = F) + scale_fill_brewer (palette = "Dark2") +
theme_classic () + geom_boxplot (width = 0.1, fill = "white")
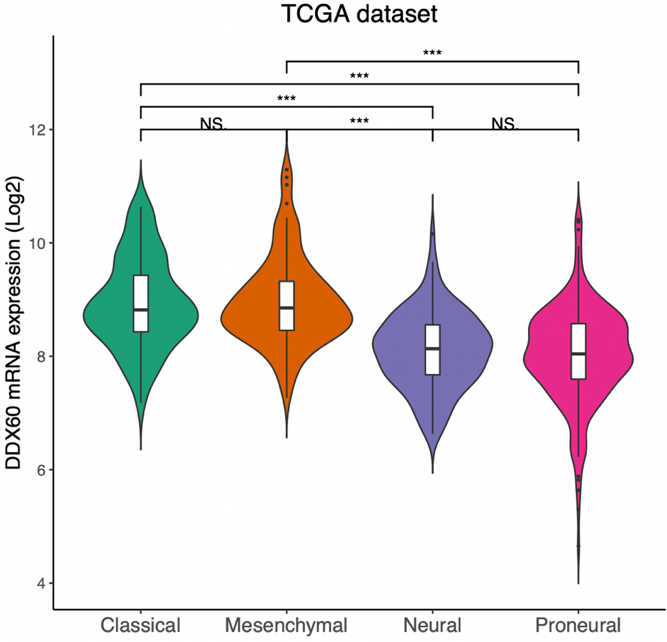
图片来源:文献截图
Figure 2
证明了目标基因在正常组和肿瘤组之间的表达存在显著差异,接下来就要证明这个目标基因是坏的,即高表达该基因的肿瘤病人生存期短,预后不良。
生存曲线就是有力证明!
想要绘制 P 值最小,即生存差异最显著的生存曲线, cut off 取值最为关键。
常用的 cut off 值一般有中位数,四分位数等,但如果想要寻找最佳 cut off 值,也可以通过 R 语言「surv_cutpoint」包来计算。
直接拿代码说事~
cut_GBM_TCGA <- surv_cutpoint (TCGA_GBM, time = "survival", event = "status", variables = c ("gene"))
一行代码搞定,最佳 cut off 值立现!
然后就需要将连续性变量按照计算好的 cut off 转为二分类变量,代码如下:
cut2_GBM_TCGA <- surv_categorize (cut_GBM_TCGA)
最后,一起来优雅地绘制生存曲线~
fit.TCGA_GBM <- survfit (Surv (survival, status) ~DDX60, data = cut2_ GBM_TCGA)
ggsurvplot (fit.TCGA_GBM)
出图如下:
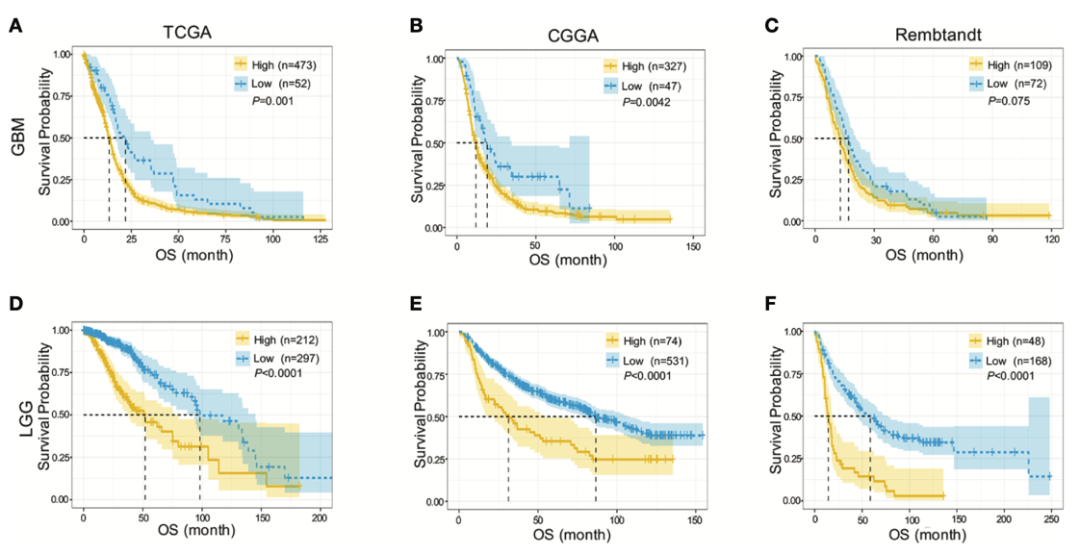
图片来源:文献截图
根据不同数据库信息,微调代码的数据框,6 张生存曲线美图分分钟搞定!
这里笔者也要提醒大家,因为本目标基因在脑肿瘤中的生存差异比较显著,因此绘制了 95% 区间的阴影线(就是蓝色、黄色的范围)毫无违和感。
如果你的目标基因画出来阴影重合比较多甚至相互交叉,建议还是去掉这个阴影吧~
Figure 3
这是一张 nomogram 图,词典翻译是列线图。
简单来说,就是将每个影响肿瘤患者预后的指标量化,并给出一个分值,将这些分值相加得到一个总分,最后根据总分对应的点,预测肿瘤患者 3 年 5 年、甚至 10 年后仍然存活的百分率。
这里的 3 年、5 年甚至 10 年都是可以自己规定的,也可以换成 3 个月、10 个月等。当然这也不是拍拍脑袋随便规定,而是要根据具体肿瘤的生存期来设置。
比如胶质母细胞瘤,脑肿瘤中最为恶性的肿瘤,其 5 年生存期不足 10%,那么这里规定 1 年、3 年即可,规定 10 年就毫无意义,图也会很难看。
Nomogram 学问还很多,一张信息量够大够好的 nomogram 图本身就可发一篇 paper,可以自己去 PubMed 一下~
好啦,这里还是纯干货,上代码。
这个代码稍稍有点复杂,比把大象放冰箱还要多一步,可以分成……4 步,我们来详细说一下:
1、打包
dd <- datadist (TCGA)
options (datadist = "dd")
2、生成函数
f <- cph (Surv (survival, status) ~ gene + Grade+ Age+ IDH.status, x = TRUE, y = TRUE, surv. = TRUE, data = TCGA, time.inc = 36)
surv <- Survival(f)
3、建立 nomogram
nomogram <- nomogram (f, fun = list (function(x) surv (36, x), function(x) surv(60, x)),
lp = FALSE, funlabel = c ("Risk of Death","3-Year Survival", "5-Year Survival"), maxscale = 100, fun.at = c (1, 0.9, 0.8, 0.7, 0.6, 0.5,0.4,0.3,0.2,0.1,0))
4、将建立好的 nomogram 绘制出来
plot (nomogram,
col.conf = c (5, 0.3),
conf.space = c(.08,.2), label.every = 1,
xfrac = 0.3, cex.axis = 0.7, cex.var = 0.9,
tcl = -0.15, lmgp = 0.05)
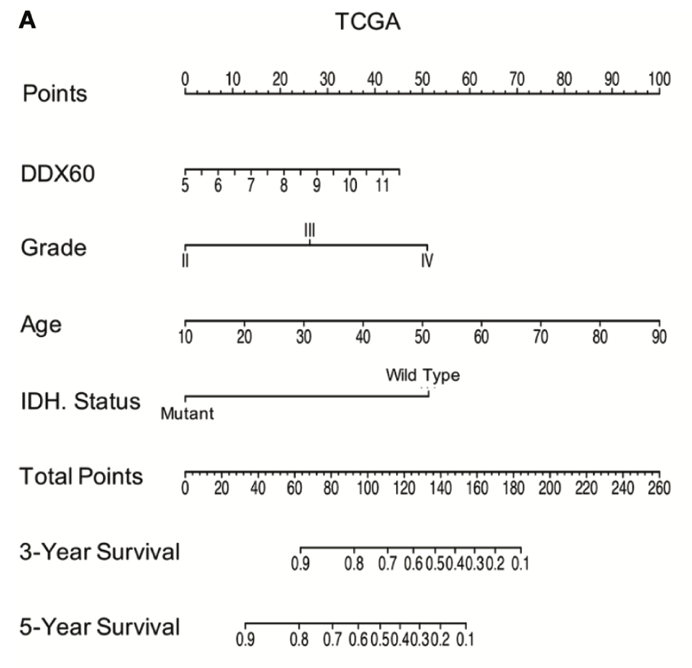
图片来源:文献截图
nomogram 绘制好之后,再进一步对其预测效果进行内部数据和外部数据的一致性和区分度的验证,绘制 calibration 图即可。
Figure 4
有现象,下一步就是探索机制了。
机制探索方式多样,基本的可以用 GO、KEGG;升级一点的,可以用 GSEA;图漂亮些的,还可以用 STRING、Cytoscope 等。
本文首先找出了 775 个与目标基因关系密切的基因,进行 GO、KEGG 分析,这一步可以用 DAVID 网站的在线工具得到数据,然后用 Excle 绘图,也可以直接用 R 语言绘图,分分钟秒杀 Excle。
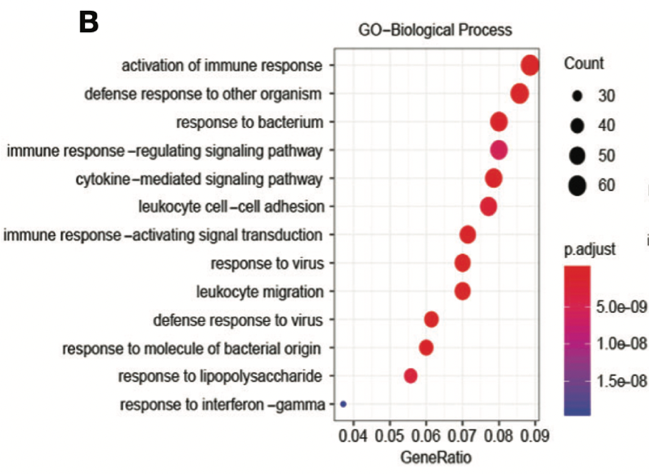
图片来源:文献截图
至于 GSEA 分析方法,网上保姆级教程比比皆是,一搜即可,不再赘述~
Figure 5 & Figure 6
这是两个 ssGSVA 分析,分别验证了目标基因与炎症反应和免疫应答密切相关。
啥是 ssGSVA?
这里我们不去绕那些非人话的名词解释,简单来说,假如我们已知目标基因和另一基因的表达量,用 pearson 检验即可得知两个基因的相关性 r 值,那么 ssGSVA 即是验证目标基因和一群基因的相关性。
如何应用?比如本文验证目标基因与炎症反应的相关性,就需要通过文献查找炎症反应包括哪些通路,如 IgG、HCK 等。
再查找这些通路上包含哪些基因,将这些基因的表达量综合,得到这个通路的整体的表达情况,再分析目标基因与这个通路的相关性。
说起来好像有点复杂,其实 R 代码只需一行:
Tmatrix <- gsva (Tt, list, method = 'ssgsea', kcdf = 'Gaussian', abs.ranking = TRUE)
然后画图,画热图还是相关性矩阵图还不是随你~
本文是都画了的,嘿嘿。
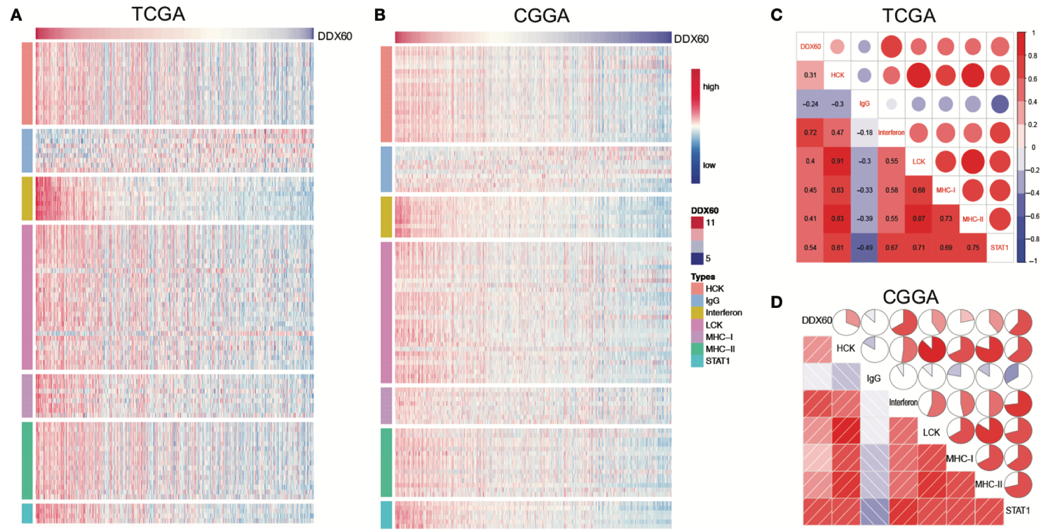
图片来源:文献截图
Figure 7
得到了这么好的结果,如何进行临床应用呢?
本文最后验证了目标基因和胶质瘤常用免疫检查位点的相关性,画了一个 circos 图,是不是像朵花一样,哈哈。
代码也如此简单~
chordDiagram (figure, symmetric = TRUE,
col = col_fun,
grid.col = 1:6,
link.sort = FALSE)
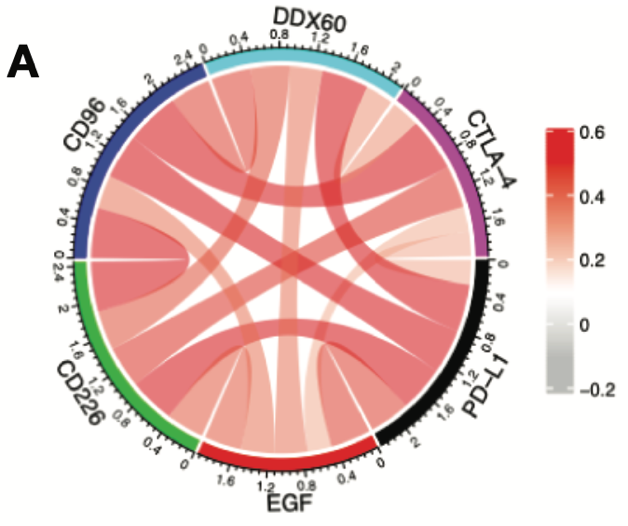
图片来源:文献截图
以上就是本文总体的生信研究思路及分析方法,get 到了吗?
一起来试试吧!你的下一篇生信 paper 正在向你招手!