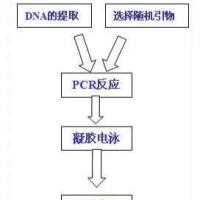
植物RAPD分析技术
互联网
2165
RAPD(Randomly Amplified Polymorphic DNA),意为随机扩增多态性DNA,是利用PCR技术进行随机扩增,把扩增的DNA片段进行琼脂糖凝胶电泳,根据DNA条带的多态性来反应模板DNA序列上的多态性。RAPD同AFLP、SSR等分子标记一样都基于PCR扩增,只是RAPD分析只需要一个引物,其引物长度一般为10个核甘酸,其序列是随机的,不同核甘酸序列的引物均有商品出售。
单引物扩增是通过一个引物在两条DNA互补链上的随机配对来完成。由于基因组DNA的差异,使引物与模板的结合位点及这些位点之间的距离不同,使PCR扩增后的片段大小表现出多态性。在基因组DNA分子内可能存在或长或短被间隔开的颠倒重复序列,如果这些序列与引物能碱基配对,则在两条单链上就会出现该引物的结合位点,结合位点的数量取决于这种重复序列的多少。不同DNA分子中这种颠倒重复序列间隔的长短不同,扩增条带的大小就不同,即表现出多态性。
10碱基引物理论上有410 种组合,不同引物检测的模板序列不同,用足够多的引物可覆盖基因组全序列,检测位点多,可以在完全不知道分子 生物 学背景的情况下对基因组进行分析,是研究植物系谱关系、起源与进化、遗传多样性及分子标记辅助育种的简单易行的方法;此外,外源基因转化后,转基因植株与非转基因植株相比,外源基因插入的部位导致基因组DNA序列不同或重排,当引物适宜时,扩增条带的长短就会不同,RAPD技术可以检测出对照植株和转化植株PCR产物带型的区别。
由于随机引物短,与模板结合的退火温度低,易出现碱基错配,因此在实验中要严格操作程序及条件,使RAPD分析的结果较为稳定。因其费用较低,方便易行,模板DNA用量少,不需要同位素,安全性好,继RFLP之后得到广泛应用;且操作上比AFLP、SSR等分子标记较为简便易行,因此,多数种类的栽培植物在资源多样性、分子标记等方面都有大量RAPD的研究报道。
本实验目的是学习RAPD技术的原理,掌握RAPD的操作及分析方法。
一、试材及用具
试材:植物基因组DNA。
用具:PCR仪,1μL、10μL、20μL、100μL微量移液器及吸管,PCR管(200μL或500μL)及管架,1.5 mL 离心管 及管架,离心机,记号笔,磁力搅拌器,高压灭菌锅,电泳仪,电泳槽,电炉,三角瓶,紫外透射仪、凝胶成像系统。
药品:10核苷随机引物、dNTP、琼脂糖、耐热DNA聚合酶[TagDNA聚合酶,与酶一起的还有两种药品,分别是10× PCR Buffer(500 mmol/LKCL、pH 9.0的100 mmol/L Tris-Cl、1.0%Tritonx⎯100)和25 mmol/L MgCl2。
其他的药品和缓冲液有:
10 mg/mL的溴化乙锭(EB)贮存液:在100 mL蒸馏水中加入1g EB,在磁力搅拌器上搅拌数h,使其完全溶解,转至棕色瓶中避光4 ℃保存。EB是强诱变剂,称量时要戴手套和口罩。一旦接触了EB,立即用大量水冲洗。
1× TAE电泳缓冲液:50× TAE浓缩贮存液含Tris碱242g;冰醋酸57.1 mL;pH 8.0的0.5 moL/L EDTA 100 mL;加600 mL蒸馏水,在磁力搅拌器上搅拌,最后加蒸馏水定容至1000 mL。
6× 凝胶加样缓冲液:0.25%溴酚蓝,0.25%二甲苯青FF,40%(W/V)蔗糖水溶液,4℃保存。
二、方法步骤
(一) 高压灭菌
把所用的吸头、PCR管、 离心管 等用具在高压灭菌锅中121℃(约1.1 kg/cm2 )高压灭菌30 min。
(二)PCR反应物的混合
所用反应体系为:25μL反应体系中,10× PCR Buffer2.5 μL,2.0 mmol/L MgCl2 ,0.2 mmol/L dNTP,,各引物0.1 μmol/L,TaqDNA酶1.5 U,各品种DNA模板浓度为20~50 ng,加重蒸水(dd H2O)补充到25 μL。
按照该反应体系,先在1.5 mL离心管中冰浴混合除模板以外的各成分,充分混匀,稍离心,分装到各PCR管中,然后把各品种的DNA模板分别加入到各管中,盖严管盖,在管外壁上做好标记,稍离心,放入PCR仪中。
(三)PCR扩增
PCR反应在DNA扩增仪中进行,所用程序为,94℃预变性4 min,94℃变性30 s,36℃退火30 s,72℃2 min,44个循环后72℃延伸10 min。反应结束后, 取出管子,置4℃冰箱中保存。
注意问题。在进行大量扩增前需对该物种确定扩增的优化体系,尤其是没有文献报道过的植物种类,如Mg2+ 适宜的浓度,TaqDNA酶的用量,退火温度等,各因子设计出梯度,通过检测扩增带的稳定性和亮度来确定优化的反应体系。
(四)琼脂糖电泳检测扩增产物
制胶。根据PCR反应管的数量和电泳槽的大小估算琼脂糖胶的体积,称1.6~2.0 %的琼脂糖溶解于1×TAE电泳缓冲液中,在电炉上加热使琼脂糖充分溶解,待溶液温度降到60℃,加入EB,使EB的终浓度为1 μg/mL,摇匀,倒入带有梳子的胶床中,避免产生气泡,室温下约30 min胶自然凝固。
点样。取出扩增DNA的管子,取7 μL扩增产物与2μL加样缓冲液混合;轻轻拔出琼脂糖胶上的梳子,把琼脂糖胶放入电泳槽中,倒入1× TAE电泳缓冲液使液面高出胶面约1 cm; 移液器 插上10 μL吸管吸入DNA样品后,尖端插入加样孔底部,缓慢把DNA样品加入到点样孔中。
电泳。加样完毕后,正确连接电泳槽和电源,黑线连阴极,红线连阳极,打开电源,正确设定电压及时间,时间的选择取决于胶的长度和电压大小。一般用不高于5v/cm的电压,电泳约2 h,待溴酚蓝离胶边约1~1.5 cm时停止电泳。
检测和照相。小心取出凝胶,置于紫外透射仪上,紫外灯下检测扩增片段的有无及分离情况,在 凝胶成像 仪上照相,图片保存于计算机中,待统计分析。
注意问题。两电极间电压不应超过5v/cm,电压越高,迁移越快,但分离效果相对较差;扩增带如果不是细亮清晰而呈形状模糊或条带粗亮相连,可能是DNA加样量太多或电压过高或 琼脂 糖浓度偏低等,在重新电泳时做适当的调整。
(五)RAPD条带分析
把RAPD每个反应重复2次。以两次重复中稳定出现的亮带作为统计对象,有电泳带记为1,无电泳带记为0,录入计算机Excel表格中。利用STATISTIC软件,采用UPGMA方法(非加权类平均法:Unweighted Pair Group Method using Arithmetric Averages)对所用品种或材料进行聚类分析。S为两样品间的遗传相似度,D为遗传距离,S=2Nxy/(Nx+Ny),D=1-S,Nxy为本两个样品共享的RAPD标记数,Nx、Ny分别为X样品和Y样品分别具有的RAPD标记数。
注意问题。对电泳结果的判读同PCR条件是否优化及引物数量是否足够等一样,会影响RAPD 结果的可信度。对电泳结果的判读主要是一些弱带的取舍问题,弱带取舍主要看其重复出现的几率,可进行重复实验;也可在难取舍的弱带位置上出现的电泳条带都不统计,这样可能丢失了一些多态性信息位点,对研究材料的系谱关系等影响不大,但当研究分子标记或构建基因连锁图谱时就不可取。
单引物扩增是通过一个引物在两条DNA互补链上的随机配对来完成。由于基因组DNA的差异,使引物与模板的结合位点及这些位点之间的距离不同,使PCR扩增后的片段大小表现出多态性。在基因组DNA分子内可能存在或长或短被间隔开的颠倒重复序列,如果这些序列与引物能碱基配对,则在两条单链上就会出现该引物的结合位点,结合位点的数量取决于这种重复序列的多少。不同DNA分子中这种颠倒重复序列间隔的长短不同,扩增条带的大小就不同,即表现出多态性。
10碱基引物理论上有410 种组合,不同引物检测的模板序列不同,用足够多的引物可覆盖基因组全序列,检测位点多,可以在完全不知道分子 生物 学背景的情况下对基因组进行分析,是研究植物系谱关系、起源与进化、遗传多样性及分子标记辅助育种的简单易行的方法;此外,外源基因转化后,转基因植株与非转基因植株相比,外源基因插入的部位导致基因组DNA序列不同或重排,当引物适宜时,扩增条带的长短就会不同,RAPD技术可以检测出对照植株和转化植株PCR产物带型的区别。
由于随机引物短,与模板结合的退火温度低,易出现碱基错配,因此在实验中要严格操作程序及条件,使RAPD分析的结果较为稳定。因其费用较低,方便易行,模板DNA用量少,不需要同位素,安全性好,继RFLP之后得到广泛应用;且操作上比AFLP、SSR等分子标记较为简便易行,因此,多数种类的栽培植物在资源多样性、分子标记等方面都有大量RAPD的研究报道。
本实验目的是学习RAPD技术的原理,掌握RAPD的操作及分析方法。
一、试材及用具
试材:植物基因组DNA。
用具:PCR仪,1μL、10μL、20μL、100μL微量移液器及吸管,PCR管(200μL或500μL)及管架,1.5 mL 离心管 及管架,离心机,记号笔,磁力搅拌器,高压灭菌锅,电泳仪,电泳槽,电炉,三角瓶,紫外透射仪、凝胶成像系统。
药品:10核苷随机引物、dNTP、琼脂糖、耐热DNA聚合酶[TagDNA聚合酶,与酶一起的还有两种药品,分别是10× PCR Buffer(500 mmol/LKCL、pH 9.0的100 mmol/L Tris-Cl、1.0%Tritonx⎯100)和25 mmol/L MgCl2。
其他的药品和缓冲液有:
10 mg/mL的溴化乙锭(EB)贮存液:在100 mL蒸馏水中加入1g EB,在磁力搅拌器上搅拌数h,使其完全溶解,转至棕色瓶中避光4 ℃保存。EB是强诱变剂,称量时要戴手套和口罩。一旦接触了EB,立即用大量水冲洗。
1× TAE电泳缓冲液:50× TAE浓缩贮存液含Tris碱242g;冰醋酸57.1 mL;pH 8.0的0.5 moL/L EDTA 100 mL;加600 mL蒸馏水,在磁力搅拌器上搅拌,最后加蒸馏水定容至1000 mL。
6× 凝胶加样缓冲液:0.25%溴酚蓝,0.25%二甲苯青FF,40%(W/V)蔗糖水溶液,4℃保存。
二、方法步骤
(一) 高压灭菌
把所用的吸头、PCR管、 离心管 等用具在高压灭菌锅中121℃(约1.1 kg/cm2 )高压灭菌30 min。
(二)PCR反应物的混合
所用反应体系为:25μL反应体系中,10× PCR Buffer2.5 μL,2.0 mmol/L MgCl2 ,0.2 mmol/L dNTP,,各引物0.1 μmol/L,TaqDNA酶1.5 U,各品种DNA模板浓度为20~50 ng,加重蒸水(dd H2O)补充到25 μL。
按照该反应体系,先在1.5 mL离心管中冰浴混合除模板以外的各成分,充分混匀,稍离心,分装到各PCR管中,然后把各品种的DNA模板分别加入到各管中,盖严管盖,在管外壁上做好标记,稍离心,放入PCR仪中。
(三)PCR扩增
PCR反应在DNA扩增仪中进行,所用程序为,94℃预变性4 min,94℃变性30 s,36℃退火30 s,72℃2 min,44个循环后72℃延伸10 min。反应结束后, 取出管子,置4℃冰箱中保存。
注意问题。在进行大量扩增前需对该物种确定扩增的优化体系,尤其是没有文献报道过的植物种类,如Mg2+ 适宜的浓度,TaqDNA酶的用量,退火温度等,各因子设计出梯度,通过检测扩增带的稳定性和亮度来确定优化的反应体系。
(四)琼脂糖电泳检测扩增产物
制胶。根据PCR反应管的数量和电泳槽的大小估算琼脂糖胶的体积,称1.6~2.0 %的琼脂糖溶解于1×TAE电泳缓冲液中,在电炉上加热使琼脂糖充分溶解,待溶液温度降到60℃,加入EB,使EB的终浓度为1 μg/mL,摇匀,倒入带有梳子的胶床中,避免产生气泡,室温下约30 min胶自然凝固。
点样。取出扩增DNA的管子,取7 μL扩增产物与2μL加样缓冲液混合;轻轻拔出琼脂糖胶上的梳子,把琼脂糖胶放入电泳槽中,倒入1× TAE电泳缓冲液使液面高出胶面约1 cm; 移液器 插上10 μL吸管吸入DNA样品后,尖端插入加样孔底部,缓慢把DNA样品加入到点样孔中。
电泳。加样完毕后,正确连接电泳槽和电源,黑线连阴极,红线连阳极,打开电源,正确设定电压及时间,时间的选择取决于胶的长度和电压大小。一般用不高于5v/cm的电压,电泳约2 h,待溴酚蓝离胶边约1~1.5 cm时停止电泳。
检测和照相。小心取出凝胶,置于紫外透射仪上,紫外灯下检测扩增片段的有无及分离情况,在 凝胶成像 仪上照相,图片保存于计算机中,待统计分析。
注意问题。两电极间电压不应超过5v/cm,电压越高,迁移越快,但分离效果相对较差;扩增带如果不是细亮清晰而呈形状模糊或条带粗亮相连,可能是DNA加样量太多或电压过高或 琼脂 糖浓度偏低等,在重新电泳时做适当的调整。
(五)RAPD条带分析
把RAPD每个反应重复2次。以两次重复中稳定出现的亮带作为统计对象,有电泳带记为1,无电泳带记为0,录入计算机Excel表格中。利用STATISTIC软件,采用UPGMA方法(非加权类平均法:Unweighted Pair Group Method using Arithmetric Averages)对所用品种或材料进行聚类分析。S为两样品间的遗传相似度,D为遗传距离,S=2Nxy/(Nx+Ny),D=1-S,Nxy为本两个样品共享的RAPD标记数,Nx、Ny分别为X样品和Y样品分别具有的RAPD标记数。
注意问题。对电泳结果的判读同PCR条件是否优化及引物数量是否足够等一样,会影响RAPD 结果的可信度。对电泳结果的判读主要是一些弱带的取舍问题,弱带取舍主要看其重复出现的几率,可进行重复实验;也可在难取舍的弱带位置上出现的电泳条带都不统计,这样可能丢失了一些多态性信息位点,对研究材料的系谱关系等影响不大,但当研究分子标记或构建基因连锁图谱时就不可取。