v468.Chapter 3 分泌型跨膜相关蛋白提纯及Wnt抑制活性
丁香园
Purification and Wnt-Inhibitory Activities of SecretedFrizzled-Related Proteins
Methods in Molecular Biology v468. chapter
Abstract
Recombinant expression of secreted Frizzled-related proteins (sFRPs) in mammalian expression systemsis a convenient source of these proteins for biological studies. Yields of protein vary; screening of clonallines for high expression is usually worthwhile. Heparin affinity chromatography is an easy step thatprovides a major enrichment, particularly for sFRP-1 and sFRP-2. Alternatively, sFRP derivatives taggedwith poly-histidine at their carboxyl termini are functional and can be readily isolated by chelating chromatography.Once purified, the proteins are stable indefinitely if stored frozen and they tolerate multiplerounds of freeze–thawing. Pre-incubation of Wnt samples with sFRP protein for 30 min at 37°C is sufficientto inhibit Wnt activity in various assays. The concentration of sFRP required to block Wnt signalingshould be determined empirically, as it will vary with the Wnt preparation and cellular context.
Key words: sFRP , Recombinant expression , Protein purification , Heparin chromatography , Chelating chromatography , Wnt
1. Introduction
Secreted Frizzled-related proteins (sFRPs) contain a Frizzled(Fzd)-type, cysteine-rich domain (CRD) that serves as a bindingsite for Wnts as well as other sFRPs and Fzds (1) . Much evidenceindicates that sFRPs can function as Wnt antagonists, presumablyby sequestering Wnts or interacting with Fzds to disruptligand-dependent receptor signaling (1 , 2) . However, additionalactivities of sFRPs have been described (2 , 3) , with one reportsuggesting that sFRP-1 itself is a ligand for Fzd-2 that regulatesaxonal extension (4) .
Although some members of the sFRP family have been isolatedfrom whole tissue or cell culture (5 , 6) , relatively small quantitieshave been obtained in this manner. There are no reportsof recombinant expression in prokaryotic systems, presumablybecause of the folding problems associated with production ofdisulfide-rich proteins in bacteria. A Fzd-type CRD has been successfullyproduced in yeast (7) ; however, we are not aware ofsimilar results for full-length sFRPs. Alternatively, mammalianexpression systems have proven to be a satisfactory source ofrecombinant, biologically active sFRP proteins (8 , 9) . Typically,yields can be optimized by screening clonally derived cell lines formaximal release of the desired protein into conditioned medium. While various purification strategies are feasible, heparin chromatographyis a straightforward procedure that does not requiresophisticated equipment and often provides purified materialsuitable for biological studies. Once isolated, sFRP proteins canbe stored frozen for use over a period of several years.
2. Materials
The following description presupposes that the reader has accessto complementary DNAs (cDNAs) encoding the sFRPs of interestor cell lines already transfected with an appropriate sFRP expressionvector. We inserted sFRP coding sequences into pcDNA3.1vectors (Invitrogen, Carlsbad, CA ), which contain a promoterfrom cytomegalovirus and antibiotic resistance elements for selection. Information about other expression vectors is available fromcommercial sources and the scientific literature. For simplicity, weoutline the scheme used to purify sFRP-1. Minor adjustments, asindicated below, also gave satisfactory results for sFRP-2. Experiencewith sFRP-3 and sFRP-4 is shared in the Notes.
2.1. Cell Culture for Conditioned Medium
1. sFRP-1/MDCK (Madin-Darby canine kidney) and sFRP-2/MDCK clonally derived, transfectant cell lines.
2. Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO;Invitrogen), with and without 10% (v/v) fetal calf serum(Biosource; Invitrogen).
3. Phosphate-buffered saline (PBS): 1.54 mM KH 2 PO 4 , 155.17mM NaCl, and 2.71 mM NaHPO 4 , pH 7.2.
4. 0.4-μm Filter.
2.2. Heparin Chromatography
1. Hi-Trap heparin affinity column (1.0-mL bed volume, GEHealthcare, Piscataway, NJ).
2. Peristaltic pump (for instance, Econo Pump, Bio-Rad,Hercules, CA).
3. Solution A: 0.05 M sodium phosphate, pH 7.4 (4×). Prepare0.2 M stock sodium phosphate monobasic solution(
13.8 g NaH 2 PO 4 ·H 2 O dissolved in 0.5 L distilled H 2 O)and 0.2 M stock sodium phosphate dibasic solution (
53.65 g Na 2 HPO 4 ·7H 2 O or 28.4 g of the anhydrous form dissolved in1 L distilled H 2 O); combine 57 mL of the former with 243mL of the latter.
4. Solution B: 0.05 M sodium phosphate, pH 7.4, and 2.0 MNaCl. Dissolve 116.8 g NaCl in 1 L Solution A.
5. Elution buffers: NaCl stepwise elution buffers are generatedby combining Solutions A and B in various ratios (for instance,5% Solution B for 0.1 M NaCl; 15% Solution B for 0.3 MNaCl; 25% Solution B for 0.5 M NaCl; 35% Solution B for 0.7M NaCl; 50% solution B for 1.0 M NaCl; and 60% solution Bfor 1.2 M NaCl).
2.3. Chelating Chromatography
1. Hi-Trap chelating affinity column (1.0-mL bed volume, GEHealthcare).
2. 0.1 M nickel sulfate (NiSO 4 ).
3. Equilibration solution: 0.05 M NaPO 4 and 0.01 M imidazole,pH 7.4.
4. Elution solutions: 0.05 M NaPO 4 and 0.05 M imidazole, pH7.4; 0.05 M NaPO 4 and 0.1 M imidazole, pH 7.4.
2.4. Sodium Dodecyl Sulfate (SDS)-Polyacrylamide Gel Electrophoresis (PAGE)
1. Mini-gel electrophoresis apparatus and power supply.
2. Pre-poured SDS-polyacrylamide mini-gels (Bio-Rad or Invitrogen);store refrigerated.
3. Running buffer: 25 mM Tris, 192 mM glycine, and 0.1 %(w/v) SDS. Store at room temperature.
4. Pre-stained molecular mass markers: Kaleidoscope markers(Bio-Rad).
5. Coomassie Brilliant Blue (CBB) staining solution: 0.025 %(w/v) CBB (Pierce), 40 % (v/v) methanol, and 7 % (v/v)acetic acid. Dissolve CBB in methanol by stirring, followed byfiltration. Then add acetic acid and bring to final volume withdistilled water.
6. Destaining solution: 30% (v/v) methanol and 10% (v/v) acetic acid.
7. Gel drying solution: 50% (v/v) methanol and 10% (v/v) acetic acid.
8. Cellophane wrap.
2.5. Immunoblotting
1. Mini-gel transfer apparatus to match electrophoresis equipment.
2. Transfer buffer: 25 mM Tris, 192 mM glycine, and 10% (v/v)methanol, pH 8.3.
3. Gel blot paper.
4. Polyvinylidene difluoride (PVDF) membrane (Millipore,Billerica, MA).
5. Methanol.
6. Tris-buffered saline (TBS): 10 mM Tris-HCl and 137 mMNaCl, pH 7.4; add 0.025% (w/v) sodium azide for longtermstorage at room temperature.
7. Blocking solution: 5% (w/v) non-fat dry milk in 0.05% (v/v)Tween-20 in TBS (T-TBS).
8. Primary antibodies to sFRPs: we generated multiple rabbit polyclonalsFRP-1 antisera directed against either the full-lengthprotein or peptide segments; alternatively, one can use commerciallyavailable reagents such as a goat polyclonal antiserum(sc-7425; Santa Cruz Biotechnology, Santa Cruz, CA).
9. Secondary antibody to use with goat polyclonal reagent: donkeyanti-goat IgG-horseradish peroxidase (HRP) (sc-2056;Santa Cruz Biotechnology).
10. Secondary antibody to use with rabbit polyclonal reagent:
donkey anti-rabbit IgG-HRP (#NA934V, GE Healthcare).
11. Enhanced chemiluminescent reagents (Femto Super Signal;Pierce, Rockford, IL).
12. X-Omat film (Kodak, Rochester, NY).
2.6. Wnt Activity Assays
1. b-catenin primary antibody: clone 4 (#610153; BD Biosciences,San Jose, CA).
2. Dishevelled (Dvl)-2 primary antibody: clone 10B5 (sc-8026;Santa Cruz Biotechnology).
3. Dvl-3 primary antibody: clone 4D3 (sc-8027; Santa CruzBiotechnology).
4. Secondary antibody to use with mouse monoclonal primaryantibodies: sheep anti-mouse IgG-HRP (#NA931V, GEHealthcare).
5. L929 fibroblasts (ATCC, Manassas, VA), RIMM-18 (10) andHEK293 cells (ATCC); Wnt-3a/L929 transfectant (ATCCline, conditioned medium routinely contains serum; we generatedanother Wnt-3a/L929 transfectant to collect serumfreeconditioned medium (11) ).
3. Methods
When expressing recombinant proteins, the choice of vector hasa major impact on purification strategy, yield, and utility of theproduct. Vectors that link a poly-histidine tag to the recombinant protein typically simplify purification by enabling chelating (nickel)chromatography to serve as a major enrichment step. In addition,an epitope tag facilitates monitoring of the protein during purification,if antibody reagents that recognize the native sequenceare not available. Epitope-tagged reagents are particularly usefulfor co-precipitation experiments, as an antibody to the foreignepitope is less likely to interfere with protein–protein interactionsmediated by the native sequence. Moreover, incorporation of thesame epitope into a series of recombinant proteins provides a wayto normalize their concentrations when only partially purifiedpreparations are available. However, the addition of an epitopewould be problematic if it disrupted protein folding and/orinteractions with other molecules.
With regard to sFRPs, heparin chromatography is an efficientpurification step, decreasing the benefit of a poly-histidine tag,and antibodies to the native proteins are commercially available. Therefore, we will describe a method for purification of recombinantproteins that lack additional sequence. Nonetheless, it isworth noting that sFRPs with tags linked to their carboxyl terminiretain functional activity (8 , 12) . Considering the potential applicationsof these derivatives, we also include a protocol for theirisolation by chelating chromatography. For recombinant expressionof sFRPs, we made a serendipitous observation that MDCKcells released a surprisingly large amount of sFRP-1 and sFRP-2into their culture fluid (yielding milligrams per liter quantities ofpurified material) (see Note 1 ). Moreover, MDCK cells are hardyand form extremely adherent monolayers, allowing for the harvestingof multiple rounds of conditioned serum-free media. Thefollowing description outlines the protocol we developed withthe MDCK recombinant expression system (see Note 2 ).
3.1. Preparation of Conditioned Medium for Protein Purification
1. Grow sFRP-1/MDCK transfectants in T175 flasks until monolayersare confluent (see Notes 3 and 4 ). After washing withPBS, maintain the cells in 25 or 30 mL of serum-free DMEM,and collect conditioned media every 3 days for five to sevenharvests. MDCK monolayers can be cycled from serum-free toserum-containing DMEM for 3-day intervals to prolong cellviability and protein synthesis (after aspirating serum-containingmedium, rinse monolayers with PBS before adding serum-freemedium for conditioning).
2. Clarify conditioned medium by centrifugation at 10,000× gfor 10 min at 4°C and filtration (see Note 5 ).
3. Place samples on dry ice for rapid freezing, and store them at–20°C or lower temperature for subsequent purification.
3.2. Heparin Chromatography
1. Thaw conditioned medium and clarify by centrifugation at10,000× g for 10 min at 4°C, followed by filtration.
2. Equilibrate heparin resin with 10 column volumes of equilibrationsolution (0.05 M phosphate buffer, pH 7.4, and 0.1M NaCl). For samples derived from as much as 2 L of conditionedmedium, use a 1-mL bed volume and a flow rate of 1mL/min. Chromatography can be performed either in a coldroom or at room temperature.
3. Load sample on resin and collect flow through (fraction thatis not retained on resin) for subsequent immunoblotting (see Note 6 ).
4. Wash resin with equilibration solution for ~10 column volumes,or until optical density of effluent is near backgroundof equilibration buffer (see Note 7 ).
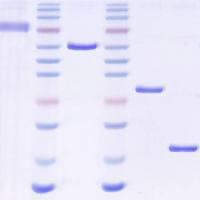
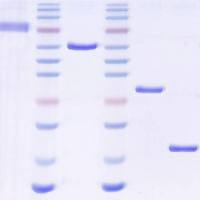
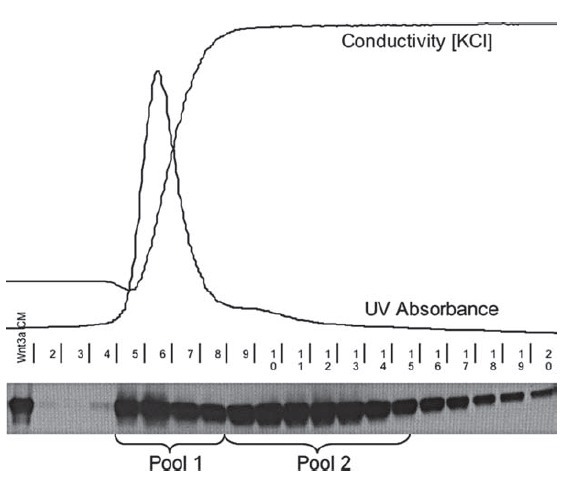
5. Elute retained protein with solutions containing stepwiseincreases in NaCl concentration. For sFRP-1, elute sequentiallywith 0.05 M phosphate buffer, pH 7.4, solutions containing0.3, 0.7, 1.0, and 1.2 M NaCl; 5–10 column volumes per step,and collect fractions corresponding to half or one columnvolume at the peak (0.5 or 1.0 mL). sFRP-1 is recovered with1.2 M NaCl (Fig. 3.1 ). For sFRP-2, elute with a slightly modified set of solutions, containing 0.3, 0.5, 0.8, and 1.0 M NaCl. sFRP-2 emerges with 0.8 M NaCl (Fig. 3.2 ) (see Note 8 ).
Fig. 3.1. Purification of recombinant sFRP-1. A Concentrated conditioned medium from sFRP-1/MDCK cells was applied toHi-Trap heparin resin (bed volume, 1 mL). Samples were eluted with increasing NaCl concentration ( dashed line ) and proteincontent was determined by monitoring optical density at 280 nm ( solid line ). Fractions are indicated on the horizontal axis.The asterisk indicates the peak containing sFRP-1. B Equal volumes (10 μL) of selected fractions eluted with 1.2 M NaClwere analyzed by 12% SDS-PAGE, and visualized by staining with CBB. S: starting material; F: flow through.
Fig. 3.2. Purification of recombinant sFRP-2. A Concentrated conditioned medium fromsFRP-2/MDCK cells was applied to Hi-Trap heparin resin (bed volume, 1 mL). Sampleswere eluted with increasing NaCl concentration ( dashed line ) and protein content wasdetermined by monitoring optical density at 280 nm ( solid line ). Fractions are indicatedon the horizontal axis. The asterisk indicates the peak containing sFRP-2. B Equal volumes(10 μL) of selected fractions eluted with 0.8 M NaCl were analyzed by 12% SDSPAGE,and visualized by staining with CBB. S: starting material.
6. Remove aliquots of fractions expected to contain sFRP proteinfor analysis by immunoblotting and protein staining (see below). Snap-freeze the remainder of these fractions and store them in a freezer. Subsequently, peak fractions can be dividedinto aliquots and refrozen. Once the purity of peak fractions isconfirmed, protein concentration can be estimated from opticaldensity (sFRP-1 and sFRP-2 extinction coefficients at 280nm are 0.968 and 0.871, respectively, for 1 mg/mL solutionsat neutral pH) (see Note 9 ) .3.3. Chelating Affinity Chromatography
1. As with heparin chromatography, freshly prepared or thawedmedium (concentrated or unconcentrated) should be clarifiedby centrifugation and filtration to minimize the likelihood offouling the resin when the sample is applied.
2. Wash chelating resin (1.0 mL) in a column with 5.0 mL ofwater (flow rate of 1.0 mL/min).
3. Charge resin with 0.5 mL of 0.1 M NiSO 4 .
4. Wash resin again with 5.0 mL distilled water.
5. Equilibrate resin with 5–10 mL of 0.05 M phosphate bufferwith 0.01 M imidazole, pH 7.4.
6. Load sample on resin, then wash with 5–10 mL of equilibrationbuffer.
7. Elute protein with eluants containing stepwise increases inimidazole; typically use buffers that have 0.05 and 0.10 Mimidazole, with sFRPs eluting at the higher concentration.
8. Analyze and store protein as indicated above for heparin chromatography.
3.4. SDS-PAGE, Followed by Protein Staining
1. Combine a standard-size aliquot (typically 5–10 μL) of elutedfractions with Laemmli sample buffer, and boil at 100°C for10 min to denature proteins.
2. Load samples in adjacent lanes of an SDS polyacrylamide gel,along with a lane containing a set of molecular mass standards.
Typically we use a pre-poured 12% or 4–20% polyacrylamidemini-gel.
3. After clamping the gel into the electrophoresis apparatus, addTris–glycine–SDS running buffer into the upper and lowerreservoirs.
4. Turn on the power supply, setting at constant voltage (140 V),and run for ~1.5 h at room temperature, until the dye frontreaches the bottom of the gel.
5. After electrophoresis, transfer the gel to a clean glass tray andrinse the gel 3× 5 min with 100–200 mL of ultrapure water.
6. Replace water with CBB staining solution, and gently rock thetray (either manually or with a platform rocker). Maximal stainingintensity usually is achieved within 15–30 min, althoughthe gel can be stained overnight (see Notes 10 and 11 ).
7. After staining/destaining has been completed, it is advisableto dry the gel for scanning and/or long-term storage. Aftermultiple washes with ultrapure water using the rocker, incubatethe gel in gel drying solution for 15–20 min in a closedcontainer on the rocker.
8. Prepare two square pieces of cellophane (each larger than thegel) and immerse them, one at a time, in gel-drying solutionfor 15–20 sec; ensure that both sides of each sheet are thoroughlywetted (wear gloves during this procedure).
9. Place the side of the gel-drying frame with holes at the cornersonto a flat surface, and center one of the sheets of prewettedcellophane on top of it.
10. Lay the gel on the cellophane, so that the gel is positionedinside the boundaries of the frame. Make sure no bubbles aretrapped between the cellophane and the gel.
11. Place the second piece of cellophane over the gel, again avoidany bubbles between gel and cellophane. Eliminate any wrinklesby smoothly rubbing the assembly with a gloved hand.
12. Align the other side of the gel-drying frame such that the cornerpins fit into the holes in the bottom side, and the positionplastic clamps at the four corners to secure the assembly.
13. Put the assembly into an upright, vertical position and let itremain standing until the gel is completely dry (12–36 h).
14. Remove the cellophane/gel sandwich from the frame, trimaway the excess cellophane, and store the sandwich betweenthe pages of a notebook under light pressure for ~2 days. Thegel now should remain flat, suitable for scanning and longtermstorage.
3.5. Immunoblotting
1. Remove the gel cassette from the electrophoresis unit andrinse it with distilled water.
2. Briefly submerge the gel in chilled transfer buffer.
3. Cut out two rectangular pieces of gel blot paper, each exceedingthe length and width of the gel by ~1.5 cm; cut out aportion of polyvinylidene fluoride (PVDF) membrane thatclosely matches the size of the gel.
4. Wet the PVDF membrane in 100% methanol for 10 sec, thenimmerse the membrane in distilled water; soak the paper andtwo foam sheets in transfer buffer.
5. Assemble a multi-layered sandwich of components in thetransfer cassette: wet foam sheet, paper, PVDF membrane,gel, paper, and second wet foam sheet; remove any bubblesfrom this assembly.
6. Place this cassette in the transfer apparatus, with the membranebetween the gel and the anode (this orientation is essential to ensure transfer of proteins onto membrane). Fillthe tank with transfer buffer, and put the lid on the tank.
7. Turn on the power supply, and run transfer at constant current(0.4 A) for 1 h. Pre-stained molecular weight markersshould be clearly visible on the membrane.
8. Incubate the membrane in blocking solution for 1 h at roomtemperature on a platform rocker.
9. Remove blocking solution, and incubate the membrane in primaryantibody solution (use goat anti-sFRP-1 reagent at a 1:500dilution) overnight (~16 h) at 4°C on a rocking platform.
10. Remove primary antibody solution, and wash membrane 4×4 min with TTBS at room temperature.
11. After the last wash, incubate the membrane in solution containingHRP–secondary antibody conjugate (donkey anti-goatIgG-HRP at a 1:1,000 dilution) for 1 h at room temperature;follow with 4× 4 min washes in TTBS.
12. Combine peroxide buffer and enhancer solutions for electrochemiluminescence(ECL) detection just prior to use for
maximal effectiveness. Pour the ECL mixture over the entiremembrane, and rock it manually to ensure even coverage.
After 1 min, place the membrane on a paper towel to removeexcess solution and then transfer the membrane into a plasticsheet protector securely fastened in a film cassette.
13. In a dark room equipped with a safe light, place X-ray film ontop of the membrane in the sheet protector and expose forvarying intervals, typically ranging from several seconds to5–10 min.
3.6. Bioassays of sFRP Effects on Wnt Activity
1. The protocol will depend on the bioassay, but the key pointis to pre-incubate the Wnt sample with the sFRP preparation.
Typically, incubate Wnt and sFRP for 30 min at 37°C prior toaddition to cells. A pilot dose–response titration of sFRP concentrationis advisable, as the effective dose will vary dependingon multiple experimental parameters (such as cell type,Wnt family member, and extracellular matrix).
2. Examples of sFRP inhibition of Wnt-3a-dependent, b-cateninstabilization and Dvl phosphorylation/mobility shift inSDS-PAGE are illustrated in Figs. 3.3 and 3.4 , respectively. In these experiments, confluent monolayers of the indicatedcells were incubated in serum-free medium overnight, followedby addition of serum-free conditioned medium fromparental or Wnt-3a-expressing L929 cells. After the specifiedtime, cells were lysed and whole cell lysates immunoblottedwith antibodies to b-catenin (1:1,000), Dvl-2 (1:500), orDvl-3 (1:500). The secondary antibody reagent was diluted at1:5,000. Other aspects of the immunoblotting protocol wereas described above.
Fig. 3.3. Dose-dependent inhibition of Wnt-3a-dependent, b-catenin stabilization inL929 cells by purified sFRP-1 and sFRP-2. After growth in serum-containing medium,L cells were incubated overnight in serum-free medium and then treated for 6 h withserum-free conditioned medium from parental L cells ( L ) or Wnt-3a/L cell transfectants( Wnt-3a ). The Wnt-3a medium had been pre-incubated with varying concentrations ofsFRPs for 30 min at 37°C before addition to cells. Cell extracts were resolved by 10%SDS-PAGE and immunoblotted ( IB ) with primary antibody directed against b-catenin.
Fig. 3.4. sFRP-1 regulation of Wnt-dependent changes in Dvl-2 ( A ) and Dvl-3 ( B ) mobility/cross-reactivity in western blots. Serum-starved RIMM-18 and HEK293 cells weretreated for 3 h either with conditioned medium from parental L cells ( L ) or Wnt-3a/L celltransfectants. Varying concentrations of sFRP-1 had been added to the Wnt-3a medium30 min before incubation with cells. Cell extracts were processed for immunoblotting( IB ) with a monoclonal antibody directed against Dvl-2 that exhibits reduced cross-reactivityfollowing Wnt-dependent, Dvl-2 phosphorylation (13) , or an antibody that recognizesDvl-3. sFRP-1 blocked the activity of added, and potentially endogenous Wnt .
4. Notes
1. To obtain clonal cell lines, we seeded the sFRP/MDCK masscultures at a 1:50,000 dilution in collagen-coated wells andsubsequently transferred colonies to culture dishes. Serumfreeconditioned media were collected from several clones, andequivalent amounts of total protein were analyzed by immunoblotanalysis for expression of sFRP. Depending on the levelof expression and antibody sensitivity, the media might have tobe concentrated with a device such as a Centricon-10 microconcentrator(Millipore) prior to immunoblotting.
2. Other commonly used mammalian expression systems haveparticular advantages. For instance, HEK293 can be adaptedfor growth in suspension, which facilitates large-scale production.
Chinese hamster ovary (CHO) cells transfected with avector encoding dihydrofolate reductase as well as the proteinof interest will undergo methotrexate-dependent amplificationof integrated plasmid copy number, increasing the synthesisof recombinant protein. The latter was used to producesFRP-3 (9) . Both cell lines are amenable to clonal selection byplating cells at sparse density and/or use of cloning cylinder.
3. T-flasks and culture plates are convenient containers forcell growth, suitable for small or intermediate production.
For larger scale operations, other devices such as cell factories(multilevel surfaces) or bioreactors for cells adapted forgrowth in suspension or on microcarriers (beads) would bebetter options.
4. Transfected cells were selected and routinely passaged inmedium containing Geneticin at 500 μg/mL (prior to transfection,one should titrate parental cells with varying concentrationsof the selective agent to determine a concentration thatwill efficiently kill cells). Omit antibiotic from cell cultures tobe used for collection of conditioned medium; selection isnot necessary at this point, and the antibiotic might interferewith bioassays of crude medium.
5. For intermediate preparations (1–2 L), concentrate clarifiedmedium ~40-fold by ultrafiltration in a stirred chamberapparatus (Amicon M2000) under nitrogen gas pressure,using a YM membrane with either a 10- or 3-kDa molecularmass cutoff. For larger preparations (several liters or more),conditioned medium can be concentrated by tangential flowultrafiltration with a Pellicon system (Millipore). Like unconcentratedmedia, concentrated samples can be snap-frozenand stored for future purification.
6. sFRP should not be present in the flow-through fraction. Ifdetected there, that would indicate the capacity of the resinhad been exceeded or there were a problem in resin equilibrationor sample preparation. For additional capacity, link Hi-Trapcolumns in series, or use a 5-mL column.
7. Ideally, chromatography would be performed with a systemthat includes a pump, on-line spectrophotometer operatingin the ultraviolet range, and a fraction collector. However, asophisticated chromatography system is not required. Accessto an ultraviolet spectrophotometer for periodic reading ofoptical density is desirable to optimize the timing of changesin elution buffers. However, once a routine is established, astandard elution protocol can be used without concomitantmonitoring of optical density.
8. For both preparations, the resin should be washed with 0.05M phosphate buffer, pH 7.4, and 2.0 M NaCl to removeresidual protein. The resin can be re-equilibrated with severalcolumn volumes of equilibration solution and stored in arefrigerator for future use. Inclusion of 0.02% (w/v) sodiumazide is prudent to prevent microbial growth.
9. Both sFRP-3 and sFRP-4 elute from heparin resin at lowerNaCl concentrations (~0.7 and 0.5 M NaCl, respectively)where more contaminants are present (lack of purity wasexacerbated by a lower level of recombinant expression bysFRP-3/MDCK and sFRP-4/MDCK transfectants). Therefore,additional chromatography is required to obtain homogeneouspreparations.
10. If CBB is too intense, the gel can be destained to improveclarity. Decant the staining solution, replace it with destainingsolution, and let the gel stir on a rocker until the backgroundis transparent and the bands well visualized. Thenwash the gel 3–5 times with water for 1–2 h prior to dryingthe gel.
11. Silver stain is more sensitive than CBB, and may detect tracecontaminants not observed with the latter. However, the relativeintensity of protein staining is more variable with silverstain than CBB.
Acknowledgments
This research was supported by the Intramural Research Programof the National Institutes of Health, National Cancer Institute.
References
1. Kawano, Y. and Kypta, R. (2003) Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 116 , 2627–2634.
2. Rubin, J.S., Barshishat-Kupper, M., Feroze-Merzoug, F., and Xi, Z.F. (2006)Secreted WNT antagonists as tumor suppressors: pro and con. Front. Biosci. 11 , 2093–2105.
3. Lee, H.X., Ambrosio, A.L., Reversade, B., and De Robertis, E.M. (2006) Embryonic dorsal-ventral signaling: secreted frizzledrelatedproteins as inhibitors of tolloid proteinases. Cell 124 , 147–159.
4. Rodriguez, J., Esteve, P., Weinl, C., Ruiz,J.M., Fermin, Y., Trousse, F., et al. (2005)SFRP1 regulates the growth of retinal ganglioncell axons through the Fz2 receptor. Nat. Neurosci. 8 , 1301–1309.
5. Hoang, B., Moos, M., Vukicevic, S., and Luyten, F.P. (1996) Primary structure and tissue distribution of Frzb, a novel protein related to Drosophila frizzled, suggest a role in human skeletal morphogenesis. J. Biol. Chem . 271 , 26131–26137.
6. Finch, P.W., He., X., Kelley, M.J., Uren, A.,Schaudies, R.P., Popescu, N.C., et al. (1997) Purification and molecular cloning of a secreted, Frizzled-related antagonist ofWnt action. Proc. Natl. Acad. Sci. U.S.A. 94 , 6770–6775.
7. Roszmusz, E., Patthy, A., Trexler, M., andPatthy, L. (2001) Localization of disulfidebonds in the Frizzled module of Ror1 receptor tyrosine kinase. J. Biol. Chem. 276 , 18485–18490.
8. Üren, A., Reichsman, F., Anest, V., Taylor,W.G., Muraiso, K., Bottaro, D.P., et al. (2000) Secreted Frizzled-related protein-1 binds directly to Wingless and is a biphasic modulator of Wnt signaling. J. Biol. Chem.275 , 4374–4382.
9. Dann, C.E., Hsieh, J.-C., Rattner, A.Sharma, D., Nathans, J., and Leahy, D.J. (2001) Insights into Wnt binding and signallingfrom the structures of two Frizzledcysteine-rich domains. Nature 412 , 86–90.
10. Levashova, Z.B., Plisov, S.Y., and Perantoni,A.O. (2003) Conditionally immortalizedcell line of inducible metanephric mesenchyme. Kidney Int. 63 , 2075–2087.
11. Endo, Y., Wolf, V., Muraiso, K., Kamijo, K., Soon, L., ?ren, A., et al. (2005) Wnt- 3a-dependent cell motility involves RhoA activation and is specifically regulated by Dishevelled-2. J. Biol. Chem . 280 , 777–786.
12. Bafico, A., Gazit, A., Pramila, T., Finch, P.W.,Yaniv, A., and Aaronson, S.A. (1999) Interactionof Frizzled-related protein (FRP)with Wnt ligands and the Frizzled receptor suggests alternative mechanisms for FRP inhibition of Wnt signaling. J. Biol. Chem . 274 , 16180–16187.
13. Gonzales-Sancho, J.M., Brennan, K.R., Castelo-Soccio, L.A., and Brown, A.M.C. (2004)Wnt proteins induce Dishevelled phosphorylationvia an LRP5/6-independent mechanism,irrespective of their ability to stabilize b-catenin. Mol. Cell. Biol . 24 , 4757–4768.