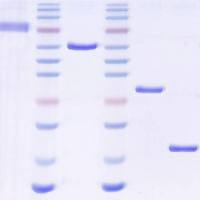
v468 chapter 5 糖原合成酶激酶-3 的抑制
丁香园
5575
Inhibition of Glycogen Synthase Kinase-3
Methods in Molecular Biology v468. chapter 5Abstract
There are two homologous forms of glycogen synthase kinase (GSK)-3, GSK-3a and GSK-3b, whichplay overlapping roles in the regulation of Wnt, Hedgehog, and insulin pathways, as well as the activationof nuclear factor (NF)-kB-mediated gene transcription. These signaling pathways regulate gene transcription,cell cycle, apoptosis, inflammation, glucose metabolism, stem-cell renewal, and differentiation.
More than 50 GSK-3 inhibitors representing a wide range of chemical structures have already been identified,and their utility in the treatment of type II diabetes mellitus, Alzheimer’s disease, bipolar diseases, cancer, andother human pathologies is currently being investigated. Here, we discuss two methods of GSK-3 inhibition,which can be used to determine the involvement of GSK-3 in a cellular process of interest.
Key words: Glycogen synthase kinase-3 , b-catenin , glycogen synthase.
1. Introduction
Glycogen synthase kinase (GSK)-3 is a serine/threonine proteinkinase which was first described as a component of insulin-mediatedsignaling pathway (1) . There are two homologous mammalianisoforms, encoded by different genes, GSK-3a and GSK-3b, sharing97% sequence similarity within their kinase catalytic domains(2) . In contrast to most kinases, GSK-3 is inactivated by phosphorylation(2) . GSK-3 is active in nonstimulated cells, whereasother kinases require cell stimulation for activation (3) . GSK-3has been shown to phosphorylate numerous physiological substrates,including metabolic enzymes and transcription factors (2) .Recently, GSK-3 has emerged as an attractive therapeutic targetfor the treatment of neurodegenerative diseases, noninsulin-dependentdiabetes mellitus, inflammation, and cancer (4 – 7) .
There are different pools of GSK-3 in the cell. The pool ofGSK-3 participating in Wnt signaling is part of a multiproteincomplex known as the b-catenin destruction complex, whichincludes b-catenin, axin, and the tumor suppressor, adenomatouspolyposis coli (APC) protein (3) . In the absence of Wnt signaling,GSK-3 targets b-catenin for ubiquitin-mediated proteosomaldegradation by phosphorylation of b-catenin, axin, and APC (3) .
Phosphorylation of APC by GSK-3 facilitates the interaction ofb-catenin and APC (8) , whereas phosphorylation of axin by GSK-3stabilizes this protein (9) . Exposure of cells to Wnt ligands leadsto b-catenin accumulation, most likely through disruption of theinteraction of axin with GSK-3 (3) , although the exact mechanismstill needs to be clarified.
Of note, the mechanism of GSK-3 inhibition by insulin signalingis different from that of Wnt signaling (10) . Insulin receptorsignaling leads to the activation of the serine/threonine kinaseAkt, which will phosphorylate GSK-3 at specific serine residues(Ser21 in GSK-3a and Ser9 in GSK-3b), leading to the inactivationof GSK-3 kinase activity (10) . However, this phosphorylationand inactivation of GSK-3 by Akt does not result in b-cateninnuclear accumulation (10) . These data indicate that the insulin/Akt signaling pathway does not affect the b-catenin destructioncomplex, but only affects a pool of GSK-3 that is involved in theregulation of glycogen synthase (4) . Accordingly, Wnt signalingdoes not lead to GSK-3 phosphorylation at Ser21/Ser9 (10) .
Although GSK-3b has been the primary focus of mostresearch, it is clear that GSK-3a and GSK-3b have some overlappingsubstrates. In fact, it has been demonstrated that tissuesfrom GSK-3b-deficient mice do not show accumulation of b-catenin(11) . In GSK-3b-deficient mouse cells, GSK-3a was also foundprecipitated with axin (11) . These data indicate that GSK-3aand GSK-3b can substitute for each other in the regulation ofb-catenin. Thus, both GSK-3a and GSK-3b need to be inhibitedto decrease b-catenin phosphorylation (Fig. 5.1a ) and induce b-cateninnuclear accumulation and transcriptional activity.
1.1. GSK-3 Inhibitors
Currently, more than 50 inhibitors of GSK-3 have been identifiedand this list is growing. Chemical structure, potency, and otherfeatures of GSK-3 inhibitors have been extensively reviewed (12 –14) . Due to the high amino acid sequence homology of GSK-3aand GSK-3b kinase domains, no pharmacological inhibitors ofGSK-3 have been identified that can differentiate between thetwo isoforms. Thus, care should be taken when using GSK-3inhibitors to assign a role for GSK-3 isoforms in specific cellularfunctions. In fact, it is advisable to use GSK-3 inhibitors alongwith genetic models that disrupt GSK-3 signaling (e.g., genetargetedcells such as mouse embryonic fibroblasts [MEFs] lacking GSK-3a or GSK-3b, or RNA interference techniques) when examining the involvement of GSK-3 isoforms in the regulationof specific cellular processes.

1.1.1. Inorganic Elements as Inhibitors of GSK-3
Lithium was the first discovered GSK-3 inhibitor (15) . Despite the factthat millimolar concentrations (20–30 mM) of lithium are requiredto inhibit GSK-3 in cells, it has been used broadly as a pharmacologicalinhibitor of GSK-3 in vitro and in vivo (13) . Lithium inhibitsGSK-3 through an unknown mechanism, although some hypotheseshave been suggested. The first hypothesis proposes lithium as amagnesium (Mg 2+ )-competitive inhibition of GSK-3 (16) , whereasthe second suggests lithium inhibitor by potassium deprivation (17) . Lithium is a nonspecific GSK-3 inhibitor and has alternative targets,e.g., inositol–phosphate phosphatases. Physiological concentrationsof zinc ions have also been shown to directly inhibit GSK-3 in vitroin an ATP-noncompetitive manner (18) . It has been shown thattreatment of HEK 293 cells with 20 μM ZnCl 2 for 2 h enhancedglycogen synthase activity twofold and increased cytoplasmicb-catenin levels (18) .
1.1.2. Small Molecule Inhibitors of GSK-3
Small molecule inhibitors of GSK-3 consist of two groups: ATPcompetitive and ATP noncompetitive. Among ATP-competitiveinhibitors of GSK-3 are indirubins (19) , paullones (20) , anilinomaleimides(21) , and other small molecules (22) . However,these inhibitors are not GSK-3 specific and they also inhibit cyclin-dependent kinases (CDKs), due to the highly related aminoacid sequence of the GSK-3 and CDK kinase domains (19) .
Accordingly, numerous CDK inhibitors are very potent GSK-3 inhibitors (19) . Recently, AR-A014418 has been reported asan ATP-competitive, potent, and specific inhibitor of GSK-3as determined by in vitro kinase assays toward cdk2, cdk5, and25 other kinases (23) . Currently thiadiazolidinone (TDZD) isthe only ATP-noncompetitive inhibitor of GSK-3 (24) . While theexact mechanism by which TDZD inhibits GSK-3 kinase activityis unclear, it has been suggested that the negative charge onthe TDZD heterocycle recognizes the oxyanion-binding site ofthe enzyme (24) , thereby blocking its ability to recognize prephosphorylatedsubstrates. The last and most specific methodto inhibit cellular GSK-3 kinase activity is depletion of GSK-3expression by RNA interference using small interfering RNAs(siRNAs) or short hairpin RNA (shRNA) targeting vectors specificfor each isoform (Fig. 5.1b ) (25) .
Although the inhibition of GSK-3 kinase activity by a certaininhibitor could be measured by in vitro kinase assay, we suggestWestern blotting as a safer, faster, and easier way to measurethe effect of an inhibitor on GSK-3 kinase activity. This can beachieved through the analysis of the phosphorylation state ofGSK-3 substrates, e.g., glycogen synthase and b-catenin. ActiveGSK-3 phosphorylates b-catenin at Ser33/37 and glycogen synthaseat Ser641/645. Commercial availability of antibodies thatrecognize phosphorylated forms of b-catenin (Ser33/37) andglycogen synthase (Ser641/645) provide straightforward assayprotocols using immunoblotting.
2. Materials
2.1. Cell Culture and Lysis
1. MiaPaCa2 and Panc04.03 pancreatic cancer cells are maintainedin DMEM and RPMI-1640 (Gibco/BRL, Bethesda, MD),respectively, supplemented with 10% fetal bovine serum (FBS;HyClone, Ogden, UT) and L -glutamine (Gibco/BRL).
2. Phosphate-buffered saline (PBS) containing 0.5 M ethylenediaminetetraacetic acid (EDTA).
3. Solution of trypsin (0.25%) containing 1 mM EDTA (Gibco/BRL).
4. Western lysis buffer for cell lysis: 10 mM Tris-HCl, pH 7.4,50 mM NaCl, 5 mM EDTA, 50 mM NaF, 30 mM Na 4 P 2 O 7 ,and 1% (v/v) Triton-X100.
5. Protease inhibitors and phosphatase inhibitor are added fresheach time with the following final concentrations: 10 μg/mLaprotinin, 10 μg/mL leupeptin, 1 mM phenylmethanesulfonylfluoride (PMSF), and 1 mM sodium orthovanadate.
6. Teflon cell scrapers (Fisher, Pittsburgh, PA ).
7. 2× SDS sample buffer for electrophoresis: 0.0375 M Tris-HCl, pH 6.5, 8% (w/v) SDS, 10% (v/v) glycerol, 5% (v/v)b-mercaptoethanol, and 0.003% (w/v) bromophenol blue.
2.2. Sodium Dodecyl Sulfate (SDS) Polyacrylamide Gel Electrophoresis (PAGE)
1. Separating buffer (4×): 1.5 M Tris-HCl, pH 8.7, and 0.4%(w/v) SDS. Store at room temperature (RT).
2. Stacking buffer (4×): 0.5 M Tris-HCl, pH 6.8, and 0.4%(w/v) SDS. Store at RT.
3. 30% acrylamide/bis solution (37.5:1 with 2.6% C) andN , N , N , N ¢-tetramethyl-ethylenediamine (TEMED; Bio-Rad,Hercules, CA).
4. Ammonium persulfate: prepare 10% (w/v) solution in waterand freeze aliquots at –20°C.
5. Water-saturated isobutanol. Store at RT.
6. Running buffer (10×): 250 mM Tris, 1.92 mM glycine, and1% (w/v) SDS. Store at RT.
7. Prestained molecular weight markers: Rainbow marker (GEHealthcare, Buckinghamshire, UK).
2.3. Immunoblotting
1. Transfer buffer (10×): 250 mM Tris-Base and 1.92 M glycine.
Store at RT.
2. Polyvinylidene difluoride (PVDF) membrane from Millipore(Bedford, MA) and gel blot paper from ISCBioExpress(Kaysville and UT).
3. Tris-buffered saline (TBS) with Tween (TBS-T): Prepare10× stock with 1.37 M NaCl, 27 mM KCl, 250 mM Tris-HCl, pH 7.4, and 1% (v/v) Tween-20.
4. Blocking buffer: 4% (w/v) bovine serum albumin (BSA) and0.1% (w/v) sodium azide in TBS.
5. Primary antibody dilution buffer: TBS-T supplemented with2% (w/v) BSA.
6. Antibodies: Phospho-b-catenin Ser33/Ser37/Thr41, phospho-glycogen synthase Ser641, and glycogen synthase (CellSignaling Technologies, Beverly, MA); b-catenin and GSK-3b(BD PharMingen, San Diego, CA); GSK-3α (Upstate, LakePlacid, NY); and b-actin (Sigma, St. Louis, MO).
7. Secondary antibodies: anti-mouse and anti-rabbit IgG conjugatedto horseradish peroxidase (Santa Cruz Biotechnology,Santa Cruz, CA).
8. Enhanced chemiluminescent (ECL) reagents from Pierce(Rockford, IL).
2.4. Stripping Blots
1. Stripping buffer: 7 M Guanidine HCl in water. Store at RT.
2. Wash buffer: TBS-T.
2.5. Transfection of Cells with Silencing shGSK-3a/b Vectors
1. GSK-3a-and GSK-3b-specific targeting shRNA vectorswere generated as previously described (26) using the targetsequences 5?-TTGTGAGGCTGAGATACTT-3? and 5?-
GATTATACCTCTAGTATAG-3?, respectively.
2. Electro Square Porator ECM830 (BTX; Genetronics Inc.,San Diego, CA).
3. Electroporation cuvettes with 4-mm gap (VWR ScientificProducts, West Chester, PA).
2.6. GSK-3 Inhibitor AR-A014418
3. Methods
3.1. Preparation of Samples for Assay of GSK-3 Activity by Western Blotting
1. Cells are treated with AR-A014418 or other GSK-3 inhibitorsaccording to the protocol and the medium is thenremoved by aspiration. Place dishes on ice and wash with 2mL of ice-cold PBS.
2. Add 0.5–1 mL of Western lysis buffer to each 10-cm plate,scrape cells, and transfer to a 1.5-mL microcentrifuge tube.
Incubate tubes on ice for 15 min.
3. Clear cell lysate by centrifuging samples at 18,000× g in arefrigerated tabletop microcentrifuge for 15 min at 4°C.
Transfer supernatant to a fresh microcentrifuge tube.
4. Measure proteins concentration by the Bradford method.
5. Mix 50 μg of proteins with 30 μL of 2× SDS-gel samplebuffer and then boil for 5 min.
3.2. Sds-page
1. These instructions assume the use of a Hoeffer SE-600 gelsystem.
2. Prepare a 1.5-mm thick, 10% gel by mixing 8.0 mL of 4×separating buffer with 13.0 mL of water, 10.5 mL of acrylamide/bis solution, 100 μL of ammonium persulfate solution, and 20 μL of TEMED. Pour the separating gel,and overlay with water-saturated isobutanol.
3. Pour off the isobutanol and rinse the top of the gel twicewith water.
4. Prepare the stacking gel by mixing 1.3 mL of 4× stackingbuffer with 3.0 mL of water, 0.8 mL of acrylamide/bis solution,30 μL of ammonium persulfate solution, and 10 μL ofTEMED. Pour the stack and insert the comb.
5. Add the running buffer to the upper and lower chambersof the gel unit and load the samples. Complete the assembly ofthe gel unit and run gel overnight at 8 mA.
3.3. Western Blotting
1. Once gel running is complete, disassemble the gel unit.
Place the separating gel on top of the PVDF membrane submergedon top of the blot paper. Another sheet of paper iswetted in the transfer buffer and laid on top of the gel. Placethe cassette into the transfer tank.
2. Transfer is accomplished at 90 V for 45 min in a transfer tankwith a connection to a refrigerated/circulating water bathmaintaining a temperature of 10–15°C.
3. After transfer, incubate the membrane in 50 mL blockingbuffer for 1 h at RT on a rocking platform.
4. Next, incubate the membrane with 30 mL of primary antibodydilution buffer containing one of the following antibodies andincubate as indicated: phospho-b-catenin (dilution 1:1,000;overnight), phospho-glycogen synthase (dilution 1:1,000; 1 hat RT), GSK-3a (dilution 1:1,000; 1 h at RT), GSK-3b (dilution1:1,000; 1 h at RT), or b-actin (dilution 1:10,000; 1 h at RT).
5. Wash the membrane three times for 5 min each in TBS-T.
6. Incubate the membrane for 1 h at RT with 30 mL of TBS-Tcontaining anti-mouse or anti-rabbit IgG conjugated tohorseradish peroxidase at a dilution of 1:5,000.
7. Wash the membrane three times for 5 min each in TBS-T.
8. Incubate the membrane with 5 mL of each enhanced chemiluminescentreagent for 5 min at RT, wrap in plastic wrap, andvisualize on X-ray film. An example result is shown in Fig. 5.1 .
3.4. Stripping and Reprobing Blots for Total b-Catenin and Glycogen Synthase
1. Incubate the membrane for 30 min at RT with 30 mL ofstripping buffer. The PVDF membrane will turn opaque.
2. Rinse the membrane in water.
3. Wash the membrane three times for 10 min each in TBS-T.
4. Reprobe the membrane with b-catenin (dilution 1:1,000;1 h at RT) or glycogen synthase (dilution 1:1,000; 1 h atRT) antibodies according to protocol described above. Anexample result is shown in Fig. 5.1b .
3.5. Transfection of Cells with Silencing shGSK-3a/b Vectors
1. Collect Panc04.03 cells from 10-cm culture dishes in the midtolate-logarithmic phase of growth. First, wash the plate with2 mL of PBS-EDTA solution and remove by aspiration. Next,add 1 mL of trypsin and incubate at 37°C for 2–3 min.
2. To the trypsinized cells, add 10 mL of complete RPMImedium, transfer to a 15-mL snapcap polypropylene tube,and centrifuge at 500× g at 4°C for 5 min.
3. Resuspend the cell pellet in the RPMI medium and measurethe cell number using a hemocytometer.
4. Collect the cells by centrifugation, as described in step 3,and resuspend them in serum-free RPMI medium at RT at aconcentration of 5×10 6 cells/mL.
5. Transfer a 400-μL aliquot of the cell suspension into a sterile1.5-mL Eppendorf tube.
6. Add 30 μg of plasmid DNA into the 1.5-mL Eppendorftube. Gently mix the cells and DNA by pipetting the solutionup and down. Keep mixture for 10 min at RT.
7. Transfer the cell–DNA mixture to an electroporation cuvetteand place in the holder for the electroporator. Discharge thedevice at 375 V for 10 ms. Remove the cuvette and keep atRT for 10 min (see Note 1 ).
8. Transfer the electroporated cells to a 10-cm tissue culturedish. Transfer the dish to a humidified incubator at 37°Cwith an atmosphere of 5–7% CO 2 .
9. The next day, observe the plates and change growth medium.
10. Prepare cell lysates 24–72 h post-transfection.
4. Notes
1. Transient transfection can be conducted by three methods: lipofection,electroporation, and polybrene plus DMSO shock. Wechose electroporation because this method is the most efficientprocedure for Panc04.03 pancreatic cancer cells, with a transfectionefficiency 90%.
Acknowledgments
This work was supported in part by the Mayo Foundation, anda Specialized Program of Research Excellence (SPORE) grant inpancreatic cancer (P50 CA102701) to DDB.
References
1. Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, WoodgettJR. (1992) Glycogen synthase kinase-3:
functions in oncogenesis and development. BiochimBiophys Acta. 1114 , 147–162.
2. Frame S, Cohen P. (2001) GSK3 takes centrestage more than 20 years after its discovery.
Biochem J. 359 , 1–16.
3. Doble BW, Woodgett JR. (2003) GSK-3:
tricks of the trade for a multi-tasking kinase.
J Cell Sci. 116 , 1175–1186.
4. Cohen P, Frame S. (2001) The renaissance ofGSK3. Nat Rev Mol Cell Biol. 2 , 769–776.
5. Cohen P, Goedert M. (2004) GSK3 inhibitors:
development and therapeutic potential.
Nat Rev Drug Discov. 3 , 479–487.
6. Ougolkov AV, Billadeau DD. (2006) TargetingGSK-3: a promising approach forcancer therapy? Future Oncol. 2 , 91–100.
7. Dugo L, Collin M, Thiemermann C. (2007)
Glycogen synthase kinase 3beta as a target
for the therapy of shock and inflammation.
Shock. 27 , 113–123.
8. Rubinfeld B, Albert I, Porfiri E, Fiol C,
Munemitsu S, Polakis P. (1996) Binding of
GSK3beta to the APC-beta-catenin complex
and regulation of complex assembly. Science.
272 , 1023–1026.
9. Yamamoto H, Kishida S, Kishida M, Ikeda S,Takada S, Kikuchi A. (1999) Phosphorylationof axin, a Wnt signal negative regulator, by
glycogen synthase kinase-3beta regulates its
stability. J Biol Chem. 274 , 10681–10684.
10. Ding VW, Chen RH, McCormick F. (2000)
Differential regulation of glycogen synthase
kinase 3beta by insulin and Wnt signaling.
J Biol Chem. 275 , 32475–32481.
11. Hoeflich KP, Luo J, Rubie EA, Tsao MS, JinO, Woodgett JR. (2000) Requirement for glycogensynthase kinase-3beta in cell survival andNF-kappaB activation. Nature. 406 , 86–90.
12. Meijer L, Flajolet M, Greengard P. (2004)Pharmacological inhibitors of glycogen synthasekinase 3. Trends Pharmacol Sci. 25 , 471–480.
13. Martinez A, Castro A, Dorronsoro I, Alonso M.
(2002) Glycogen synthase kinase 3 (GSK-3)
inhibitors as new promising drugs for diabetes,neurodegeneration, cancer, and inflammation.
Med Res Rev. 22 , 373–384.
14. Alonso M, Martinez A. (2004) GSK-3
inhibitors: discoveries and developments.
Curr Med Chem. 11 , 755–763.
15. Stambolic V, Ruel L, Woodgett JR. (1996)
Lithium inhibits glycogen synthase kinase-3
activity and mimics wingless signalling in
intact cells. Curr Biol. 6 , 1664–1668.
16. Ryves WJ, Harwood AJ. (2001) Lithium
inhibits glycogen synthase kinase-3 by competitionfor magnesium. Biochem Biophys ResCommun. 280 , 720–725.
17. Mora A, Sabio G, Gonzalez-Polo RA, et al.
(2001) Lithium inhibits caspase 3 activation
and dephosphorylation of PKB and GSK3
induced by K+ deprivation in cerebellar
granule cells. J Neurochem. 78 , 199–206.
18. Ilouz R, Kaidanovich O, Gurwitz D, Eldar-Finkelman H. (2002) Inhibition of glycogensynthase kinase-3beta by bivalent zinc ions:
insight into the insulin-mimetic action of zinc.
Biochem Biophys Res Commun. 295 , 102–106.
19. Leclerc S, Garnier M, Hoessel R, et al.
(2001) Indirubins inhibit glycogen synthase
kinase-3 beta and CDK5/p25, two protein
kinases involved in abnormal tau phosphorylationin Alzheimer’s disease. A propertycommon to most cyclin-dependent kinase
inhibitors? J Biol Chem. 276 , 251–260.
20. Leost M, Schultz C, Link A, et al. (2000)Paullones are potent inhibitors of glycogen synthasekinase-3beta and cyclin-dependent kinase5/p25. Eur J Biochem. 267 , 5983–5994.
21. Smith DG, Buffet M, Fenwick AE, et al. (2001)3-Anilino-4-arylmaleimides: potent and selectiveinhibitors of glycogen synthase kinase-3 (GSK-3).
Bioorg Med Chem Lett. 11 , 635–639.
22. Cross DA, Culbert AA, Chalmers KA, Facci L,Skaper SD, Reith AD. (2001) Selective smallmoleculeinhibitors of glycogen synthase kinase-3activity protect primary neurones from death.
J Neurochem. 77 , 94–102.
23. Bhat R, Xue Y, Berg S, et al. (2003) Structuralinsights and biological effects of glycogen synthasekinase 3-specific inhibitor AR-A014418.
J Biol Chem. 278 , 45937–45945.
24. Martinez A, Alonso M, Castro A, Perez C,
Moreno FJ. (2002) First non-ATP competitive
glycogen synthase kinase 3 beta
(GSK-3beta) inhibitors: thiadiazolidinones
(TDZD) as potential drugs for the treatment
of Alzheimer’s disease. J Med Chem.
45 , 1292–1299.
25. Ougolkov AV, Fernandez-Zapico ME,
Savoy DN, Urrutia RA, Billadeau DD.
(2005) Glycogen synthase kinase-3beta
participates in nuclear factor kappaB-mediatedgene transcription and cell survivalin pancreatic cancer cells. Cancer Res. 65 ,
2076–2081.
26. Zakaria S, Gomez TS, Savoy DN, et al. (2004)Differential regulation of TCR-mediatedgene transcription by Vav family members.
J Exp Med. 199 , 429–434.