v468 Chapter 2 Wnt活性蛋白分离及应用
丁香园
2580
Isolation and Application of Bioactive Wnt Proteins
Methods in Molecular Biology v468. chapter 2
Abstract
Wnt proteins and their signaling cascades are involved in a wide variety of developmental processes, andderegulation of this pathway is frequently associated with tumorigenesis. Unlike many other growthfactors, Wnts long eluded biochemical purification, in large part because of their hydrophobic nature,which is imparted by one or more lipid modifications (1 – 3) . Here I describe a complete protocol thatoutlines the purification process for Wnt proteins. While this protocol has not been applied to all knownWnt proteins, it has been successfully applied to the purification of a large subset of Wnts, including thevery divergent Wnt protein, Drosophila Wnt8 (Dwnt8 or WntD), indicating that this protocol is likelyapplicable to all Wnts.
Key words: Wnt , Wnt3A , b-catenin , Purification , Blue Sepharose , Immobilized metal affinitychromatography (IMAC) , Gel filtration , Heparin cation exchange .
1. Introduction
The protocol described here, based on a previous publication (3) ,outlines the purification protocol of Wnt proteins, starting with acrude and dilute Wnt sample, usually in the form of conditionedmedium (CM) (4) , to the final purified protein. In addition, protocolsto assay Wnt activity are described.
Two key observations have made the purification of Wntspossible: 1) inclusion of detergent to facilitate solubility of thehighly hydrophobic Wnt protein, and 2) fractionation over BlueSepharose, which binds Wnts with high selectivity. The purificationconsists of four chromatography steps: Blue Sepharose, immobilized metal affinity chromatography (IMAC), gel filtration,and heparin cation exchange. Throughout the purification,samples are assayed for both the presence of the Wnt protein(using a Wnt immunoblot or a Coomassie- or silver-stained gel)and activity (b-catenin stabilization or activation of Wnt reporterconstructs) to ensure optimal recovery of protein and associatedactivity.
This protocol describes the purification of Wnt3A specifically;modifications for other Wnt proteins are necessary (e.g.,elution profiles may vary; for detection use the appropriate Wntantibody; not all Wnts stabilize b-catenin; etc.). The resultingpurified product is useful in a variety of assays, including signalingstudies in established cell lines, explant manipulations, and in vivoexperiments (for examples, see refs. (3 , 5 – 15) ).
2. Materials
2.1. Production of Wnt3A CM
1. Cell line: L-Wnt3A (ATCC , Manassas, VA; ATCC# CRL-2647) (see Note 1 ).
2. Cell culture medium: Dulbecco’s minimum essential medium(DMEM) supplemented with 10% (v/v) fetal bovine serum(FBS), 1:100 dilution of penicillin–streptomycin solution with10,000 U penicillin (base)/mL and 10,000 μg streptomycin(base)/mL in 0.85% NaCl (liquid) (see Note 2 ).
3. Dulbecco’s phosphate-buffered saline (PBS).
4. Trypsin (0.25% liquid trypsin).
5. Cell culture dishes: 10- or 15-cm dishes or large surface areaculture dishes (e.g., Corning® CellSTACK®).
6. CO 2 incubator and biosafety cabinet.
7. Filter bottle, 0.5–1 L, 0.2-μm pore size (Corning or equivalent).
2.2. Preparation of Wnt3A CM for Fractionation
1. 20% (v/v) Triton X-100.
2. 1 M Tris-HCl, pH 7.5.
3. 10% (w/v) NaN3 .
4. Filter bottle, 0.5–1 L, 0.2-μm pore size (Corning or equivalent).
2.3. Fractionation of Wnt3A CM
2.3.1 Step 1: Blue Sepharose
1. Sample: 1–4 L Wnt3A CM as prepared in Section 2.2 .
2. Column: Blue Sepharose HP (Cibacron Blue F3G-A coupledto Sepharose), ~100 mL packed into an empty column with100–200 mL of bed volume.
3. Binding Buffer: 1% (w/v) CHAPS, 150 mM KCl, and 20 mMTris-HCl, pH 7.5, sterile filtered.
4. Elution Buffer: 1% (w/v) CHAPS, 1.5 M KCl, and 20 mMTris-HCl, pH 7.5, sterile filtered.
5. Syringe (30–50 mL) with 0.2-μm pore size filter.
2.3.2. Step 2: IMAC
1. Sample: Pooled Wnt3A containing fractions from Section2.3.1 .
2. HiTrap? Chelating, 1-mL column (GE Healthcare, Piscataway,NJ), loaded with Cu 2+ .
3. Binding Buffer: 0.5 M NaCl, 20 mM Tris-HCl, pH 7.5, and1% (w/v) CHAPS.
4. Elution Buffer: Binding Buffer with 100 mM imidazole, pH 7.5.
5. Syringe (10 mL) with 0.2-μm pore size filter.
2.3.3. Step 3: Gel Filtration
1. Sample: 5–10 mL pooled Wnt3A-containing fractions fromSection 2.3.2 .
2. HiLoad 26/60 Superdex 200 preparative grade.
3. Buffer: 1× PBS, 1% (w/v) CHAPS.
4. Syringe (30 mL) with 0.2-μm pore size filter.
2.3.4. Step 4: Heparin Cation Exchange
1. Sample: 20–40 mL pooled Wnt3A-containing fractions fromSection 2.3.3 .
2. 1 mL HiTrap heparin (GE Healthcare).
3. Binding Buffer: 1× PBS, 1% (w/v) CHAPS.
4. Elution Buffer: Binding Buffer, 1 M NaCl (adjust pH to thatof Binding Buffer if necessary).
5. Syringe (10 mL) with 0.2-μm pore size filter.
2.4. Assays for Wnt Proteins
2.4.1. Wnt3A Immunoblots
1. Sample: Any fraction containing the Wnt protein of interest.
2. Protein Sample Loading Dye (4×): 250 mM Tris-HCl, pH6.8, 8% (w/v) sodium dodecyl sulfate (SDS), 40% (v/v)glycerol, 20% (v/v) 2-mercaptoethanol, pinch of bromophenol blue.
3. Suitable SDS-polyacrylamide gel electrophoresis (PAGE) andtransfer setup (e.g., the BioRad Criterion Precast Gel Systemor equivalent).
4. Nitrocellulose or polyvinyl difluoride (PVDF) membrane.
5. Wnt3A antibody (available from R&D Systems, Minneapolis,MN).
6. Appropriate conjugated secondary antibody for detection.
2.4.2. b-Catenin Stabilization
1. Mouse L-cells (ATCC CCL-1.3 or CRL-2648).
2. Cell culture medium: Same as Section 2.1.2.
3. 96-well tissue culture plate (flat well).
4. Wnt3A samples (CM, fractions from Sections. 2.3 and 2.4 ).
5. PBS.
6. Lysis Buffer: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and1% (v/v) Triton X-100.
7. Suitable SDS-PAGE and transfer setup as in Section 2.4.1 .
8. Protein Sample Loading Dye (4×) as in Section 2.4.1 .
9. Nitrocellulose or PVDF membrane.
10. Mouse anti-β-catenin antibody (available from various vendors,including BD Transduction Laboratories, FranklinLakes, NJ; R&D Systems; and Santa Cruz Biotechnology,Santa Cruz, CA).
11. Appropriate conjugated secondary antibody for detection.
3. Methods
3.1. Production of Wnt3A CM
1. Grow L-Wnt3A cells to confluency.
2. Wash with warm (37°C) PBS, trypsinize, and divide theL-Wnt3A cells into plates with a surface area 20 times greaterthan the original dish(es). Use about 10 mL of cell culturemedium per 75-cm 2 surface area).
3. Incubate cells in a humidified CO 2 incubator at 37°C for4 days.
4. Remove CM and filter through 0.2-μm filter. Add fresh mediato all plates and incubate another 3 days.
5. Harvest a second batch of CM. Discard dishes. Filter thesecond batch and combine with first batch of CM. The CMmedia can be stored at 4°C for several months without appreciablereduction in activity, as assessed by b-catenin stabilizationin L-cells, over the course of 1 year. However, a sensitiveand quantitative assay may detect a change in specific activity.
3.2. Preparation of Wnt3A CM for Fractionation
1. To the filtered CM from Section 3.1 , add Triton X-100 to1% (v/v), Tris-HCl, pH 7.5, to 20 mM, and NaN 3 to 0.01%(w/v) (e.g., to 930 mL CM, add 50 mL of 20% (v/v) TritonX-100, 20 mL of 1 M Tris-HCl, pH 7.5, and 1 mL of 10%(w/v) NaN 3 ).
2. Filter through a 0.2-μm filter.
3.3. Fractionation of Wnt3A CM
This fractionation can be performed with any chromatographysetup, such as the ?kta FPLC (GE Healthcare). If such an instrumentis not available, the purification can also be performed usinga peristaltic pump that can accurately control flow rates of 1–5mL/min. The purification involves four steps: Blue Sepharose,IMAC, gel filtration, and heparin cation exchange, each of whichis described in detail below. All buffers and the sample should besterile filtered through a 0.2-μm filter (see Note 3 ).
3.3.1. Step 1: Blue Sepharose
Purpose: This step serves to recover a large fraction of Wnt3Aprotein from the CM. In an optimal experiment, the Wnt proteincan be enriched 2,000- to 2,500-fold (see Notes 4 and 5 ).
1. Column Packing: Pour a 1:1 slurry of the Blue Sepharose intoa clean and empty column, such as XK26-20 or XK50-20 (GEHealthcare). Allow resin to settle overnight; close the columnfrom the top by lowering and tightening the plunger so thatno air is trapped and no gap exists between the top of the resinbed and the plunger.
2. Column Equilibration: Using a suitable pump, wash the columnwith 2–3 column volumes (CV) of filtered and distilledwater or 20 mM Tris-HCl, pH 7.5, at a flow rate of 1–5 mL/min, being sure not to exceed the allowable back pressure forthe Blue Sepharose resin. Wash the column with 2–3 CV ofbinding buffer. If using a UV monitor, adjust it to zero. Thecolumn is now ready for sample application.
3. Sample Application: At a flow rate of 1–5 mL/min, applythe entire volume of the Wnt3A CM. The flow rate can beadjusted so that this step can be performed within the spanof a few hours or overnight (e.g., a 1 L sample can be appliedat 5 mL/min in 3 hours and 20 minutes or at 1 mL/min in16 hours and 40 minutes). Collect the flow-through material,which can be examined for the presence of Wnt proteinby immunoblotting to ensure depletion (see Note 6 ). Duringthis loading step, the UV monitor may exceed its detectionlimit, which is in part due to the high protein content of thesample (10% FBS) and in part due to the Triton X-100, anonionic detergent containing an aromatic group that exhibitsintense UV absorbance.
4. Washing Column: Wash the column with Binding Buffer untilthe UV reading has stabilized near baseline. This may takefour to five CVs. Washing can be done at a flow rate of upto 5 mL/min, as long as the maximal allowed back pressuretolerated by the resin is not exceeded.
5. Elution: Once the UV reading has established a stable baseline,start the elution by switching the buffer to ElutionBuffer. Start collecting 10-mL fractions. A large protein peak will emerge immediately coincident with the increase in saltconcentration. Continue to collect fractions for about 100 to200 mL beyond the main protein peak. Assay all fractions forWnt3A protein by immunoblotting (see Section 3.4.1 ). It isimportant to note that a large portion of the Wnt3A proteintrails behind the main protein peak. These trailing Wntfractions (Pool 2 in Fig. 2.1 ) contain very little of the otherproteins that bind to Blue Sepharose under these conditions. If this separation is not achieved and all the Wnt protein coeluteswith the main protein peak, the following fractionationsteps will serve to remove the majority of contaminating proteins. Combine the fractions containing the highest amountsof Wnt protein and sterile filter through a 0.2-μm syringe filter. Depending on the amount of starting material and thesize of the column, the total volume of pooled eluate fractionswill vary from 40 to 100 mL. Store the samples at 4°C beforeproceeding to the next step. Do not freeze the fractions.

6. Regeneration of column: Wash the column into distilled waterand then into 20% (v/v) ethanol for long-term storage. Toclean the column rigorously, wash the column in reverse flow with two to three CVs of 0.1–0.5 N NaOH. This treatmentwill remove some of the immobilized Cibacron Blue dye fromthe resin, so contact time should be minimized (see Note 4 ).
3.3.2. Step 2: IMAC
Purpose: This step serves to remove contaminating proteins andto concentrate the volume of Wnt3A-containing fractions.
1. Column Equilibration: At a flow rate of 1 mL/min, wash thecolumn with 5 mL distilled and filtered water, and then with5 mL Binding Buffer. The column is now ready for sampleapplication.
2. Sample Application: Apply the pooled and filtered fractionsfrom Step 1 at a flow rate of 1 mL/min, using a Superloop TM(GE Healthcare) or a sample pump. Collect the flow-throughmaterial. Once the entire sample has been applied, wash thecolumn with 5 to 10 CVs of Binding Buffer or until the UVabsorbance has established a stable baseline.
3. Elution: Bound protein is eluted by a combination of a stepfollowed by a linear gradient elution (see Fig. 2.2 ): 1) stepto 5% Elution Buffer/95% Binding Buffer and collect 1-mLfractions for 10 CV (note: a large protein peak should eluteas shown in Fig. 2.2 ); 2) elute with a gradient from 5% ElutionBuffer/95% Binding Buffer to 100% Elution Buffer over 10 CV and collect 1-mL fractions; 3) collect an additionalten 1-mL fractions at 100% Elution Buffer. Verify the presenceof Wnt3A by immunoblotting (see Section 3.4.1 ). Thevast majority of Wnt3A should be present in fractions collectedduring the gradient from 5 to 100% Elution Buffer. The bound Wnt protein elutes in a broad peak, totalingabout 5 mL, at a concentration of approximately 10–40 mMimidazole. If sufficient Wnt3A CM is fractionated (>1 L), aCoomassie stained gel should be sufficient to allow detectionof the Wnt protein in the eluate fractions (as shown in Fig. 2.2 ). Combine fractions containing highest amounts of Wntprotein and sterile filter through a 0.2-μm syringe filter. Thesample can be stored at 4°C before continuing with the purification. Do not freeze the fractions.

3.3.3. Step 3: Gel Filtration
Purpose: This step serves to separate low molecular weight Wntfrom high molecular weight Wnt and some contaminating proteins. Additionally, it serves to exchange the buffer from high(0.5 M) to physiological (1× PBS) salt concentrations, which isimportant to perform Step 4 of this purification.
1. Column Equilibration: At a flow rate of 1–2.5 mL/min (besure not to exceed the maximal back pressure permissible forthis column), wash the column with one CV distilled and filteredwater, then with two CV Buffer. The column is nowready for sample application.
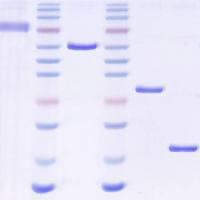
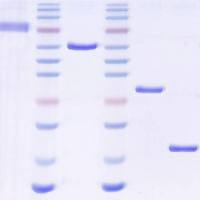
2. Sample Application and Fractionation: Load the sample usinga Superloop TM at a flow rate of 1 to 2.5 mL/min. If the sampleexceeds the maximum volume that can be efficiently fractionatedon this column (consult the vendor’s recommendationsfor details), split the sample and perform two identical fractionations. Once the entire sample has been applied, collect10-mL fractions for an entire CV. The majority of the Wntprotein should emerge at the same position as a molecularweight standard of 50 kD (see Fig. 2.3 ). Occasionally a smallamount of Wnt protein elutes with the void volume of thecolumn suggesting that it is part of a large complex or aggregate(see Note 7 ). The Wnt protein can be readily detected ona Coomassie-stained gel. Combine all Wnt-containing fractionsand pass through a 0.2-μm syringe filter.

Fig. 2.3. Gel filtration of Wnt3A and b-catenin assay. The elution peak positions ofmolecular weight standards on the Superdex 200 column are indicated at the top. Wnt3A has an approximate molecular weight of 50 kD ( top panel ). Eluate fractionswere diluted 1:100 in DMEM with 10% FBS and applied to mouse L cells for a b-cateninstabilization assay (bottom panel, see Section 3.4.2 ). Only fractions containing Wnt3Astimulate the stabilization of b-catenin.
3.3.4. Step 4: Heparin Cation ExchangePurpose: This fractionation step serves to concentrate the Wntprotein and remove remaining contaminating proteins, such asbovine serum albumin (BSA).
1. Column Equilibration: Wash the column with 5 mL distilledand filtered water at a flow rate of 1 mL/min, then with5 mL Binding Buffer. The column is now ready for sampleapplication.
2. Sample Application: Load the entire sample from Section3.3.3 and collect the flow-through material. Upon loading,wash the column with 5 to 10 CV or until UV absorbance hasestablished a stable baseline.
3. Elution: Elute the bound Wnt protein by applying a lineargradient from 0 to 100% Elution Buffer. The Wnt proteinelutes in a tight peak at approximately 200 mM NaCl (i.e., 20%Elution Buffer = PBS + 200 mM NaCl). Alternatively, theWnt protein can be eluted in a single step from 0 to 100%Elution Buffer. The maximum concentration of Wnt that can beachieved in these buffer conditions is approximately 100 μg/mL(2.5 μM for Wnt3A). The Wnt protein can be readily detectedon a Coomassie-stained gel. Combine all Wnt-containingfractions, filter through a 0.2-μm syringe filter, and store(see Note 8 ).
3.4.1 Wnt3A Immunoblotting
1. Prepare samples by combining a small amount of a Wnt samplewith 4× loading dye and water to give a final concentrationof the loading dye of 1× in a total volume of 20 to 40μL. Use the following amounts of Wnt per 20 μL sample: 1)Wnt3A CM: 1 μL; 2) Blue Sepharose fractions: 1 μL; 3)IMAC fractions: 1 μL for Wnt3A immunoblot (or 10 μL for Coomassie-stained gel); 4) gel filtration fractions: 2 μL forWnt3A immunoblot (or 15 μL for Coomassie-stained gel); 5)heparin fractions: 1 μL for Wnt3A immunoblot or Coomassie-stained gel.
2. Boil all samples for 5 minutes.
3. Perform SDS-PAGE (10% acrylamide gel) followed by transferto a suitable membrane, such as nitrocellulose or PVDFusing standard protocols.
4. Detect Wnt3A protein using a Wnt3A-specific antibody accordingto manufacturer’s recommendations.
3.4.2. b-Catenin Stabilization
1. One or 2 days prior to the assay, seed a 96-well plate with L-cells(see Note 9 ). Seeding one tenth of the cells from a confluent10-cm dish of L-cells into all 96 wells will give the desired celldensity to perform the assay. Most of the following manipulationscan be performed with 12-well multichannel pipetters.
2. Prepare samples to be tested in a separate 96-well plate: To100 μL of cell culture media, add 1–2 μL of the Wnt3Acontainingfractions (see Note 2 ).
3. Aspirate media from L-cells in 96 well plates.
4. Transfer diluted Wnt samples to the cells. For a positive control,use 100 μL of Wnt3A CM per well and for a negativecontrol use cell culture medium (or CM from L-cells preparedas described for Wnt3A CM in Section 3.1 ).
5. Incubate cells for 2 hours at 37°C in a humidified CO 2 incubator.
6. Aspirate all control and test wells and wash once with 100 μLof PBS.
7. Add 30 μL of Lysis Buffer, incubate 1–2 minutes, and transferlysates (do not try to dislodge nuclei by scraping) to tubescontaining 10 μL of Protein Sample Loading Dye.
8. Boil and resolve protein samples by SDS-polyacrylamide gelelectrophoresis.
9. Transfer protein from the gel to a suitable membrane andperform a β-catenin immunoblot as recommended by manufacturer. An example of this β-catenin stabilization assay isshown in Fig. 2.3 .
4. Notes
1. This purification can be applied to other Wnt proteins: to date,Wnt3A, 5A, 7A, 16, Wingless, and Dwnt8/WntD have beensuccessfully purified using this scheme (3 , 11) . Aside from the cells producing Wnt3A, ATCC makes available a similar cellline overexpressing Wnt5A (ATCC# CRL-2814).
2. Lowering FBS concentrations in the media will lead to a reductionof soluble Wnt3A in the CM. Since Wnt proteins areextremely hydrophobic, it is likely that the high lipid contentof serum promotes the solubility of the Wnt protein. Uponfractionation of the Wnt CM, the requirement for serum isdisplaced by the presence of detergent. If purified Wnt3A isdiluted into an aqueous buffer, either detergent or 10% FBSshould be included. In fact, dilution of purified Wnt into abuffer lacking either detergent or serum will lead to precipitationand loss of activity.
3. The purification has been successfully performed at 4°C androom temperature. However, I have not carefully examinedwhether one of these temperatures yields a superior purifiedWnt protein preparation. Given common wisdom of proteinstability and temperature, I recommend that the purificationis performed at 4°C and all samples are stored in the cold.
I recommend against freezing any samples, from the startingmaterial to the final purified protein fractions, as this mayresult in protein denaturation and precipitation.
4. While Blue Sepharose binds all the Wnts tested so far, I recommendthat a given column is dedicated to the purificationof a single Wnt protein. Wnt proteins are extremely sticky andare likely present at low levels even after washing the columnextensively.
5. During the Blue Sepharose fractionation, the detergent isexchanged from 1% Triton X-100 to 1% CHAPS. This isdone because Triton X-100 is significantly less expensive thanCHAPS. However, Triton X-100 is not a suitable detergent forthe entire purification because of its low critical micelle concentration(0.0155% w/v) relative to CHAPS (0.492–0.615%w/v) and its cellular toxicity. In addition, in the presence ofTriton X-100, Wnt3A is incorporated into high molecularweight complexes (potentially micelles) as assessed by gel filtration,and is significantly less active.
6. The concentration of Wnt3A in CM is approximately 200ng/mL. Blue Sepharose has a binding capacity of approximately0.64 mg Wnt3A protein per 100 mL Blue SepharoseHP. This estimation is derived from these observations: themaximal amount of Wnt3A CM loaded onto a 100-mL BlueSepharose column has been 4 L (~0.8 mg Wnt3A) and 80%of Wnt3A was depleted, indicating that 0.64 mg of Wnt3Awas bound. Flow-through fractions collected at the end of theloading step contained significantly more Wnt3A protein thanflow-through fractions collected at the beginning, suggesting that the column was approaching saturation with respect toWnt binding.
7. By gel filtration, the majority of Wnt3A should emerge as aprotein whose molecular size corresponds closely to its monomericor slightly larger size. A high molecular weight complexcontaining Wnt3A is often observed. This high molecularweight Wnt is most likely aggregated protein, because 1) it isless active than low molecular weight Wnt3A, 2) it cannot bereadily dissociated with detergent, and 3) it can be pelleted bycentrifugation (50,000× g ).
8. The stability and shelf life of purified Wnt protein, specificallyWnt3A, is not known. However, it is clear that purified Wntprotein is unstable and looses activity over time. I have foundthat after 6 months of storage at 4°C, only 10% activity willremain. Therefore, it is best to use the Wnt protein shortlyafter purification. Preliminary studies indicate that the purifiedWnt protein can be flash frozen in liquid nitrogen and storedat –150°C without appreciable loss in activity. While it hasnot been tested experimentally, it is likely that stored undersuch conditions, the Wnt protein will retain maximal activityindefinitely. Avoid repeated freeze–thaws by freezing downmultiple small volume aliquots. Some Wnt proteins (Wnt3a,5a, 5b, and 7a) are provided by R&D Systems in a lyophilizedform, suggesting that lyophilization does not adversely affectthe activity of the Wnt protein.
9. Mouse L cells lack cadherins and consequently have very littleto no membrane-associated β-catenin. As a result, the Wntstimulatedcytoplasmic accumulation of β-catenin is readilydetectable. If cells with cadherins are used in place of L cells,the Wnt-stimulated cytoplasmic accumulation of β-cateninmay be masked by the large amounts of β-catenin at the membrane. In this case, cells should be fractionated to separatecytosolic proteins from membrane-bound proteins (e.g., bylysis in hypotonic buffer without detergent).
References
1. Kurayoshi, M., Yamamoto, H., Izumi, S., and Kikuchi, A. (2007) Post-translational palmitoylation and glycosylation of Wnt-5a are necessary for its signalling. Biochem J . 402 , 515-523.
2. Takada, R., Satomi, Y., Kurata, T., Ueno, N.,Norioka, S., Kondoh, H., et al. (2006)Monounsaturated fatty acid modificationof Wnt protein: its role in Wnt secretion. Dev Cell , 11 , 791-801.
3. Willert, K., Brown, J. D., Danenberg, E., Duncan, A. W., Weissman, I. L., Reya, T., et al. (2003) Wnt proteins are lipid-modifiedand can act as stem cell growth factors. Nature , 423 , 448-452.
4. Shibamoto, S., Higano, K., Takada, R., Ito, F.,Takeichi, M., and Takada, S. (1998)Cytoskeletal reorganization by solubleWnt-3a protein signalling. Genes Cells , 3 ,659-670.
5. Bryja, V., Schulte, G., and Arenas, E. (2007) Wnt-3a utilizes a novel low dose and rapid pathway that does not require casein kinase 1-mediated phosphorylation of Dvl to activate beta-catenin. Cell Signal ,19 , 610-616.
6. Bryja, V., Schulte, G., Rawal, N., Grahn, A.,and Arenas, E. (2007) Wnt-5a inducesDishevelled phosphorylation and dopaminergicdifferentiation via a CK1-dependentmechanism. J Cell Sci , 120 , 586-595.
7. Gadue, P., Huber, T. L., Paddison, P. J., and Keller, G. M. (2006) Wnt and TGFbeta signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc Natl Acad Sci U S A , 103 , 16806-16811.
8. Galli, L. M., Barnes, T., Cheng, T., Acosta, L.,Anglade, A., Willert, K., et al. (2006) Differentialinhibition of Wnt-3a by Sfrp-1, Sfrp-2,and Sfrp-3. Dev Dyn , 235 , 681-690.
9. Galli, L. M., Willert, K., Nusse, R., Yablonka-Reuveni, Z., Nohno, T., Denetclaw, W.,et al. (2004) A proliferative role for Wnt-3ain chick somites. Dev Biol , 269 , 489-504.
10. Kishida, S., Yamamoto, H., and Kikuchi, A. (2004) Wnt-3a and Dvl induce neurite retraction by activating Rho-associated kinase. Mol Cell Biol , 24 , 4487-4501.
11. Mikels, A. J. and Nusse, R. (2006) PurifiedWnt5a protein activates or inhibitsbeta-catenin-TCF signaling depending onreceptor context. PLoS Biol , 4 , e115.
12. Reya, T., Duncan, A. W., Ailles, L., Domen,J., Scherer, D. C., Willert, K., et al. (2003)A role for Wnt signalling in self-renewal ofhaematopoietic stem cells. Nature , 423 ,409-414.
13. Schmitt, A. M., Shi, J., Wolf, A. M., Lu,C. C., King, L. A., and Zou, Y. (2006)Wnt-Ryk signalling mediates medial-lateralretinotectal topographic mapping. Nature ,439 , 31-37.
14. Schulte, G., Bryja, V., Rawal, N., Castelo-Branco, G., Sousa, K. M., and Arenas, E. (2005) Purified Wnt-5a increases differentiationof midbrain dopaminergic cells anddishevelled phosphorylation. J Neurochem ,92 , 1550-1553.
15. Yue, Z., Jiang, T. X., Widelitz, R. B., andChuong, C. M. (2006) Wnt3a gradientconverts radial to bilateral feather symmetryvia topological arrangement of epithelia. Proc Natl Acad Sci U S A , 103 , 951-955.