【心得】测序常见问题及分析
丁香园论坛
9540
总结了测序中经常遇到的问题及原因分析,希望对大家有帮助!
Q:测序结果中为什么找不到引物序列?
A:找不到用来测序的引物,这是正常的,因为测序的方法是荧光标记测序法,仪器通过检测 ddNTP 上的荧
光来读取所测序列,而引物本身是不被标记的,所以仪器无法检测到;
找不到所测片段的扩增引物,这种情况是因为您所采用的酶切位点离所用的测序引物距离太近,一般荧光
燃料会干扰几十个碱基的读取,这部分碱基会损失掉,损失掉的序列很可能就包含引物的序列,所以引物
的序列无法找到;
测出的引物序列是原引物序列的反向互补序列,这是因为 TA 克隆的插入没有方向性,如果插入片段是相
反的,这时就要反向互补查找引物;
测出的结果为空载体,这是因为由于某种原因导致质粒上没有插入外源片段,这时所测的为载体序列,所
以就找不到引物了;
存在单引物扩增,有一条引物的特异性不好,有多个结合位点,导致只有一条引物参与扩增。
Q:为什么测序结果中引物的序列有个别碱基不同了?
A: 这是因为引物区同样存在错配的可能,出现这种可能性有两种: 合成错误和引物区有错配
我们可以挑选同批次 2个以上的克隆进行测序验证,如果结果完全相同,应该是合成错误;如果在引物的
相同位置错误的碱基不一样那就应该是引物区有错配。
Q:哪些引物不适合作为测序引物?
A:①兼并引物:简并引物要在测序模板上有多个结合位点,直接影响测序结果;
②随机引物:如 RAPD 引物,随机引物一般都比较短,所用退火温度低,在测序反应的条件下,不能很好
地与模板结合;
③过长的引物:一般要求测序引物不大于 24bp,过长的引物在测序反应的较低的条件下容易在测序模板上
有多个结合位点,导致测序结果背景增高。另外,较长的引物纯度也将难以保证。通常用于测序的引物纯
度要在 90%以上,引物纯度低时,测序反应的背景将明显增大,直接影响到测序结果;
④有特殊标记的引物:该情况主要指荧光标记的引物。我们测序反应的四种碱基都是荧光标记的,这样,
荧光标记的引物将产生干扰。另外,其他一些有大的标记基团的引物也最好不要用于测序。引物上大的标
记基团将直接影响到 DNA 片段的迁移率,导致测序结果峰型不好或错误;
⑤不纯的引物:测序引物对纯度的要求很高,合成的引物中非全长的片段可以造成较强的背景。测序的引
物最好是经过 PAGE 胶纯化的。
Q: 用测序的方法检测点突变可靠吗?
A: 该方法可靠性不高,主要有以下两个原因。
①首先,并不清楚突变的序列与正常的序列的比例是多少。测序反应的信号强度直接与模板的量有关,如
果突变的模板所占的比例很少,将直接作为背景噪音了,很难检测出来。只有当测序反应体系中正常的和
突变的模板量比较接近时,才能较可靠地检测到突变体的存在;
②其次,在同一位置,不同碱基的信号强渡一般是不一样的。这样即使突变的模板所占的比例较高时,也
不一定能准确检测到突变的存在。另外,测序仪是设计用来测序正常的碱基序列的,软件在对扫描的结果
进行处理时, 会尽量提高主峰而将背景信号尽量压低, 以得到尽可能好的结果。 因此, 当某处出现双峰时,
测序仪一般会认为信号弱的峰为背景信号,在处理过程中,将弱的峰进一步压低,这样根部不立于突变体
的检测。因此认为,用测序的方法检测突变体的存在不是一个好的方法。
Q:DNA测序样品用什么溶液溶解比较好?
A:溶解DNA测序样品,用灭菌双蒸馏水最好。DNA 的测序反应也是 Taq酶的聚合反应,需要一个最佳的酶
的反应条件。用缓冲液溶解在进行测序反应时,缓冲液的组份会影响到测序反应,条件,造成 Taq 酶的聚
合性能下降。
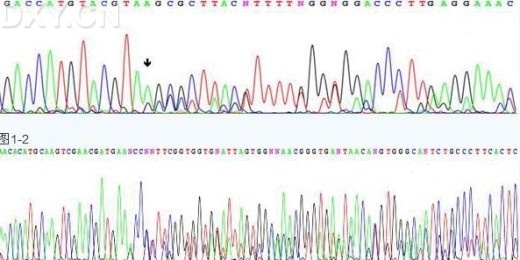
Q:测序结果中会出现双峰的现象,是什么原因造成的?
A:样品本身被污染,这常常发生在样品为质粒和菌液的情况中。当使用通用引物测序时,如果刚好和样品
中的几个质粒均能结合,那么就会出现这种情况,而且在同一位置上还可能有不止两个峰形;
样品不是单一模板,这常常发生在样品为PCR产物的情况中。由于使用的是特异性引物,因此即使模
板中有其他 DNA 的存在,产生干扰的可能性也很小。通常是由于存在两条分子量很接近,采用琼脂糖电泳
无法分开的条带,在测序时就会发生这种情况;
样品中存在两个引物结合位点,这常常发生在样品中存在重复序列的情况下。当引物恰好设计在重复
序列中时,那么在重复序列以外的部分就会出现 Two pattern 的情况;
所用的测序引物不纯, 这种情况一般发生在PCR产物测序中。 因为PCR测序一般使用的都是PCR扩增引物,
如果引物本身不纯,测序结果会出现双峰。所以我们测序的引物一般要求都是要经过 PAGE 纯化的;
测序样品序列中存在如重复序列,poly 结构,发夹结构等复杂结构导致测序信号出现乱峰。
Q:DNA片段过长,一个反应测不通怎么办?
A: 从两端同时测序,然后拼接;如果两端同时测序还是没通,这时可以在原测序结果上合适区域设计引
物步行测序,测通后拼接即可。
Q:过短的PCR 产物为什么不适于直接测序?
A:①首先,过短的 PCR产物纯化困难,一般的 PCR 产物纯化试剂盒都要求 PCR 产物片段大于 150bp,过短
的 PCR 产物纯化和准确定量都非常困难。因此我们要求用于测序的 PCR 产物一般不低于 150bp长度。
②其次,由于测序本身的限制,以一个 100bp的 PCR 产物用于测序为例,去掉两个引物的序列大约 40 到
50bp,再加上测序起始端的一些读不出的碱基,真正能够得到的有用序列不过 30~40 碱基。这么少的序
列很难向客户收费。因此,过短的 PCR 产物,只能克隆后进行测序。
Q: 我的样品上有一个杂合位点,为什么在你们的报告上看不到杂合的信号?
A:①在检查报告时,设备和我们的技术员都倾向于提供给客户一个单一的信号,所以在出现杂合的位置上
给出的信号往往是比较强的一个信号。 所以如果您的PCR样品上是存在杂合位点的, 请在测序订单上注明,
我们在修改报告时会加以注意。
②如果在您预期出现杂合信号的位置上只有单一的信号,那么我们是不会人为将其修改为杂合位点的。出
现这样的情况可能是在您的样品中杂合成份太少,信号强度不足以被检查到。
Q:我的基因序列与标准序列为什么有差别?
A: 一段基因序列经扩增后,克隆到载体中进行测序。在两个层次上可能导致序列发生变化。首先在 PCR
扩增过程中就可能产生错误。将片段克隆到载体中也有可能发生突变。其次,测序的准确率问题。ABI 公
司承诺其仪器的测序精度并没有达到 100%。由于仪器准确率的限制,在一个较长的序列中发生碱基序列错
误是难以避免的。在确认克隆无误的情况下,通过双向测序可以最大限度减少测序的错误。您如果想得到
您的最准确的序列,进行双向测序是很有必要的。
Q:测序结果中为什么找不到引物序列?
A:找不到用来测序的引物,这是正常的,因为测序的方法是荧光标记测序法,仪器通过检测 ddNTP 上的荧
光来读取所测序列,而引物本身是不被标记的,所以仪器无法检测到;
找不到所测片段的扩增引物,这种情况是因为您所采用的酶切位点离所用的测序引物距离太近,一般荧光
燃料会干扰几十个碱基的读取,这部分碱基会损失掉,损失掉的序列很可能就包含引物的序列,所以引物
的序列无法找到;
测出的引物序列是原引物序列的反向互补序列,这是因为 TA 克隆的插入没有方向性,如果插入片段是相
反的,这时就要反向互补查找引物;
测出的结果为空载体,这是因为由于某种原因导致质粒上没有插入外源片段,这时所测的为载体序列,所
以就找不到引物了;
存在单引物扩增,有一条引物的特异性不好,有多个结合位点,导致只有一条引物参与扩增。
Q:为什么测序结果中引物的序列有个别碱基不同了?
A: 这是因为引物区同样存在错配的可能,出现这种可能性有两种: 合成错误和引物区有错配
我们可以挑选同批次 2个以上的克隆进行测序验证,如果结果完全相同,应该是合成错误;如果在引物的
相同位置错误的碱基不一样那就应该是引物区有错配。
Q:哪些引物不适合作为测序引物?
A:①兼并引物:简并引物要在测序模板上有多个结合位点,直接影响测序结果;
②随机引物:如 RAPD 引物,随机引物一般都比较短,所用退火温度低,在测序反应的条件下,不能很好
地与模板结合;
③过长的引物:一般要求测序引物不大于 24bp,过长的引物在测序反应的较低的条件下容易在测序模板上
有多个结合位点,导致测序结果背景增高。另外,较长的引物纯度也将难以保证。通常用于测序的引物纯
度要在 90%以上,引物纯度低时,测序反应的背景将明显增大,直接影响到测序结果;
④有特殊标记的引物:该情况主要指荧光标记的引物。我们测序反应的四种碱基都是荧光标记的,这样,
荧光标记的引物将产生干扰。另外,其他一些有大的标记基团的引物也最好不要用于测序。引物上大的标
记基团将直接影响到 DNA 片段的迁移率,导致测序结果峰型不好或错误;
⑤不纯的引物:测序引物对纯度的要求很高,合成的引物中非全长的片段可以造成较强的背景。测序的引
物最好是经过 PAGE 胶纯化的。
Q: 用测序的方法检测点突变可靠吗?
A: 该方法可靠性不高,主要有以下两个原因。
①首先,并不清楚突变的序列与正常的序列的比例是多少。测序反应的信号强度直接与模板的量有关,如
果突变的模板所占的比例很少,将直接作为背景噪音了,很难检测出来。只有当测序反应体系中正常的和
突变的模板量比较接近时,才能较可靠地检测到突变体的存在;
②其次,在同一位置,不同碱基的信号强渡一般是不一样的。这样即使突变的模板所占的比例较高时,也
不一定能准确检测到突变的存在。另外,测序仪是设计用来测序正常的碱基序列的,软件在对扫描的结果
进行处理时, 会尽量提高主峰而将背景信号尽量压低, 以得到尽可能好的结果。 因此, 当某处出现双峰时,
测序仪一般会认为信号弱的峰为背景信号,在处理过程中,将弱的峰进一步压低,这样根部不立于突变体
的检测。因此认为,用测序的方法检测突变体的存在不是一个好的方法。
Q:DNA测序样品用什么溶液溶解比较好?
A:溶解DNA测序样品,用灭菌双蒸馏水最好。DNA 的测序反应也是 Taq酶的聚合反应,需要一个最佳的酶
的反应条件。用缓冲液溶解在进行测序反应时,缓冲液的组份会影响到测序反应,条件,造成 Taq 酶的聚
合性能下降。
Q:测序结果中会出现双峰的现象,是什么原因造成的?
A:样品本身被污染,这常常发生在样品为质粒和菌液的情况中。当使用通用引物测序时,如果刚好和样品
中的几个质粒均能结合,那么就会出现这种情况,而且在同一位置上还可能有不止两个峰形;
样品不是单一模板,这常常发生在样品为PCR产物的情况中。由于使用的是特异性引物,因此即使模
板中有其他 DNA 的存在,产生干扰的可能性也很小。通常是由于存在两条分子量很接近,采用琼脂糖电泳
无法分开的条带,在测序时就会发生这种情况;
样品中存在两个引物结合位点,这常常发生在样品中存在重复序列的情况下。当引物恰好设计在重复
序列中时,那么在重复序列以外的部分就会出现 Two pattern 的情况;
所用的测序引物不纯, 这种情况一般发生在PCR产物测序中。 因为PCR测序一般使用的都是PCR扩增引物,
如果引物本身不纯,测序结果会出现双峰。所以我们测序的引物一般要求都是要经过 PAGE 纯化的;
测序样品序列中存在如重复序列,poly 结构,发夹结构等复杂结构导致测序信号出现乱峰。
Q:DNA片段过长,一个反应测不通怎么办?
A: 从两端同时测序,然后拼接;如果两端同时测序还是没通,这时可以在原测序结果上合适区域设计引
物步行测序,测通后拼接即可。
Q:过短的PCR 产物为什么不适于直接测序?
A:①首先,过短的 PCR产物纯化困难,一般的 PCR 产物纯化试剂盒都要求 PCR 产物片段大于 150bp,过短
的 PCR 产物纯化和准确定量都非常困难。因此我们要求用于测序的 PCR 产物一般不低于 150bp长度。
②其次,由于测序本身的限制,以一个 100bp的 PCR 产物用于测序为例,去掉两个引物的序列大约 40 到
50bp,再加上测序起始端的一些读不出的碱基,真正能够得到的有用序列不过 30~40 碱基。这么少的序
列很难向客户收费。因此,过短的 PCR 产物,只能克隆后进行测序。
Q: 我的样品上有一个杂合位点,为什么在你们的报告上看不到杂合的信号?
A:①在检查报告时,设备和我们的技术员都倾向于提供给客户一个单一的信号,所以在出现杂合的位置上
给出的信号往往是比较强的一个信号。 所以如果您的PCR样品上是存在杂合位点的, 请在测序订单上注明,
我们在修改报告时会加以注意。
②如果在您预期出现杂合信号的位置上只有单一的信号,那么我们是不会人为将其修改为杂合位点的。出
现这样的情况可能是在您的样品中杂合成份太少,信号强度不足以被检查到。
Q:我的基因序列与标准序列为什么有差别?
A: 一段基因序列经扩增后,克隆到载体中进行测序。在两个层次上可能导致序列发生变化。首先在 PCR
扩增过程中就可能产生错误。将片段克隆到载体中也有可能发生突变。其次,测序的准确率问题。ABI 公
司承诺其仪器的测序精度并没有达到 100%。由于仪器准确率的限制,在一个较长的序列中发生碱基序列错
误是难以避免的。在确认克隆无误的情况下,通过双向测序可以最大限度减少测序的错误。您如果想得到
您的最准确的序列,进行双向测序是很有必要的。