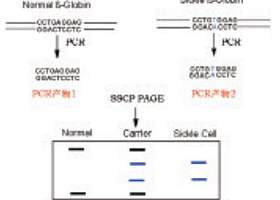
pcr-sscp法
丁香园论坛
4805
PCR-SSCP分析的基本程序为:首先PCR扩增特定靶序列,然后将扩增产物变性为单链,进行非变性聚丙烯酰胺凝胶电泳。在不含变性剂的中性聚丙烯酰胺凝胶中电泳时,DNA单链的迁移率除与DNA链的长短有关外,更主要的是取决于DNA单链所形成的构象。在非变性条件下,DNA单链可自身折叠形成具有一定空间结构的构象。这种构象由DNA单链碱基决定,其稳定性靠分子内局部顺序的相互作用(主要为氢键)来维持。相同长度的DNA单链其顺序不同,甚至单个碱基不同,所形成的构象不同,电泳迁移率也不同。PCR产物变性后,单链产物经中性聚丙烯酰胺凝胶电泳,靶DNA中含单碱基置换,或数个碱基插入或缺失等改变时,因迁移率变化会出现泳动变位,从而可将变异DNA与正常DNA区分开。由此可见,PCR-SSCP分析技术是一种DNA单链凝胶电泳技术,它根据形成不同构象的等长DNA单链在中性聚丙烯酰胺凝胶中的电泳迁移率变化来检测基因变异。该技术已被广泛用于癌基因和抗癌基因变异的检测、遗传病的致病基因分析以及基因诊断、基因制图等领域。
为了高灵敏特异性地显示SSCP分析结果,现已发展多种PCR-SSCP技术,各有其优势及适用领域。例如,可以在PCR扩增中用同位素或荧光素等标记引物或核苷,也可以在电泳后用银染或溴化乙锭染色以显示结果。这里将重点介绍最常用的放射性同位素PCR-SSCP法。在进行PCR扩增特定靶基因序列时,利用γ-32P-ATP标记引物或直接在PCR反应体系中加入α-32P-dCTP进行PCR扩增,使扩增产物带有同位素标记物,然后将扩增产物变性为单链进行非变性聚丙烯酰胺凝胶电泳,放射自显影显示结果。利用引物标记或碱基掺入法使PCR扩增产物带有同位素标记物,均可使产物信号增强几个数量级。二者相比较,前者经济、扩增特异性强,多用于大样本的检测和筛选;后者操作简便,适于一般实验室开展,可用于小样本或大样本的检测和筛查。本节仅介绍同位素标记碱基掺入法。
一、试剂准备
⒈ PCR相关试剂
⒉ α-32P-dCTP
⒊ 30%聚丙烯酰胺(29:1):丙烯酰胺29g、甲叉双丙烯酰胺 1g,溶于100ml ddH2O中,4 OC保存。
⒋ 10%过硫酸胺配制方法:1g 过硫酸胺,溶于10ml ddH2O中,4 OC保存(可用数周)。
⒌ TEMED(N,N,N,N’,N’-四甲基乙二胺)
⒍ 5×TBE缓冲液:Tris碱54g、硼酸27.5g、0.5M EDTA(pH 8.0) 20ml,加ddH2O至1000ml。
7.变性上样液:95% 甲酰胺、0.03%二甲苯青、0.05%溴酚蓝、 20mM EDTA(pH 8.0)
二、操作步骤
⒈ PCR扩增
反应总体积为10μl,在0.5ml微量离心管中加入下列反应成分:
10×buffer 1μl
dNTPmix 70μM
DNA模板 100ng
引物及Taq DNA酶 依实验设计要求按比例加入
α-32P-dCTP 0.1μl
加ddH2O至 10μl
按实验设计循环参数进行扩增,获取扩增产物。
⒉ 聚丙烯酰胺凝胶电泳
⑴ 电泳槽玻璃板的处理:用洗涤剂清洗玻璃板,自来水反复冲净洗涤剂,双蒸水冲洗3次,晾干,95%乙醇擦拭,自然干燥。用棉签沾取SigmaCote涂于玻璃的贴胶面上, 5~10min后再用软纸擦拭除去多余的SigmaCote溶液。在两块玻璃内的两侧放好衬条对齐,用固定夹夹紧两块玻璃,并用玻璃胶带封边。
⑵ 制胶:按照被分离DNA片段的大小、含量及玻璃板、衬条的大小决定凝胶的浓度与体积,一般来讲使用5%~8%的凝胶较为合适。轻轻摇匀配制的胶液于真空抽气(开始时要缓慢)除气泡,加 35μl TEMED至聚丙烯酰胺混合液中,混匀。用10ml玻璃吸管或50ml注射器吸取胶液,将玻璃模具倾斜成60O角,缓慢注入两玻璃板间的空隙中,直至灌满模具顶部。立即插入相应的点样梳,(小心勿使梳齿下带进气泡,并且不要将梳齿全部插入胶内,留约2mm梳齿于玻璃板上端,以免拔梳时把胶孔拔断)。由于凝胶在聚合过程中有回缩,所以应小心添加些胶液于梳子处,水平放置,室温聚合1hr。将封口玻璃胶带掀去,放入电泳槽,凹型玻璃贴紧电泳缓冲液槽,用大号固定夹固定住两侧。在上下电泳槽内灌入 1×TBE电泳缓冲液。小心取出点样梳,用槽内的缓冲液反复冲洗点样孔,以去除可能存在的未聚合的聚丙烯酸胺和气泡。
⑶ 扩增产物的处理:将 PCR扩增产物与变性上样液按1:5的比例加入0.5ml 微量离心管中,混匀。样品上胶前应98 o C变性10min,立即冰浴骤冷。取约3~5μl变性样品(根据点样孔的大小决定上样量),以微量加样器上样,(上样时要注意不要有气泡冲散样品,而且速度要快,时间长了样品易于扩散)。
⑷ 电泳:电泳槽接上电极(上槽接负极,下槽接正极)开启电源,根据扩增片段的大小及电泳槽和凝胶的大小,决定电泳的电压及电泳时间。通常室温下以l~5V/cm电泳。
⑸ 剥胶:电泳结束后,倒弃电泳缓冲液,取下电泳胶玻璃,用塑料楔子从玻璃板底部一角小心分开玻璃,凝胶应附着在一块玻璃上,切去凝胶左上角,作为点样顺序标记。剪一张与玻璃同样大小的滤纸,严密覆盖于凝胶上,顺一个方向将凝胶缓慢取下,用保鲜膜盖于胶面并包好,避免膜和胶之间产生气泡或皱折,将凝胶固定在X线片夹中。
⑹放射自显影:在暗室中将X线片贴于胶面,盖严片夹,用黑布将X线片夹包裹,置-70OC放射自显影。曝光时间根据同位素的强度而定,约 24h~10d。
⑺ 冲洗胶片从-70oC取出X线片夹,在暗室内迅速取出X线片,立即显影,以免使X线片上出现过多的冷凝水。(如果想再得到一张放射自显影影像,可立即在片夹中装一张新X线片并尽快放回到一70oC继续显影。如果来不及再加入新片即已出现冷凝水则应使样品凝胶和X线片夹都恢复到室温,并擦去冷凝水后再装入新的X线片)。依次按以下程序操作进行显影:
X线片显影液显影 1-5min
水洗 lmin
定影液定影 5min
流动水冲洗 15min
所有使用液的温度应为18~20OC为宜。
三、注意事项
⒈ 核酸片段的大小: 用于SSCP分析的核酸片段越小,检测的敏感性越高。对于<200bp的片段,SSCP可发现其中70%的变异;对于300bP左右的片段则只能发现其中50%的变异;而>500bp的片段,则仅能检出10%~3O%的变异,因此,<300bP,尤其是150bP左右的核酸片段更适于SSCP分析。对于大于400bp的PCR产物就需要设法进一步处理,可以用限制性酶消化PCR产物,产生小于400bP的DNA片段,再进行SSCP分析。
⒉ 游离引物: 游离引物可能同 PCR产物结合而改变其泳动率,即使游离引物量为6nM都有明显影响。因此,应尽可能除去游离引物。可以采用不对称引物扩增方法,尽可能消耗多余的引物。也可以运用过柱或磁性球方法纯化PCR产物。或者是稀释PCR产物,减少游离引物的干扰。
⒊ 低浓度变性剂: 凝胶中加入低浓度的变性剂,如5%-10%甘油、5%尿素或甲酸胺、10%二甲亚砜(DMSO)或蔗糖等有助于提高敏感性,可能是因为轻微改变单链DNA的构象,增加分子的表面积,降低单链DNA的泳动率。但有些变异序列却只能在没有甘油的凝胶中被检出。因此,对同一序列使用2~3种条件做SSCP,可能提高敏感性。
⒋ 电泳温度: 一般认为保持凝胶内温度恒定是SSCP分析最关键的因素,温度有可能直接影响DNA分子内部稳定力的形成及其所决定的单链构象,从而影响突变的检出。室温下电泳适于大多数情况,但由于在电泳时温度会升高,为确保电泳温度相对恒定,应采取以下措施:减少凝胶厚度,降低电压,有效的空气冷却或循环水冷却等。
⒌ 凝胶的长度: 可用测序板进行SSCP分析,凝胶板长度在40cm以上。
⒍ 凝胶浓度及厚度: 凝胶浓度很重要,一般使用5%~8%的凝胶,凝胶浓度不同,突变带的相对位置也不相同,如果在进行未知突变种类的SSCP分析时,最好采用两种以上凝胶浓度,这样可以提高突变种类的检出率。凝胶的厚度对SSCP分析也很重要,凝胶越厚,背景越深,在上样量较多的前提下,尽量使凝胶越薄越好。
⒎ 假阴性: 一般认为,如没有污染,PCR-SSCP分析不存在假阳性结果,但可能出现假阴性结果,后者是由于点突变引起的空间构象变化甚微,迁移率相差无几所致,尤其是点突变发生在扩增片段的两端时。如果有阳性和阴性对照,结果可以重复确定的突变带是可信的,如果没有阳性对照,应经测序来确定其是否为突变带。由于PCR-SSCP的不足之处主要是可能检出假阴性结果。应通过设置阳性对照,摸索电泳条件,假阴性结果在很大程度上是可以避免的。但对未知基因变异的检测,假阴性结果就难以百分之百地消除。
8. 结果分析: 单链凝胶电泳时,互补单链迁移率不同,一般形成两条单链带。PCR产物进行单链凝胶电泳之前,通过加热变性产生单链。如变性不彻底,残留双链亦可形成一条带.因此,PCR-SSCP分析结果至少显示三条带。但是,由于一种DNA单链有时可形成两种或多种构象,检出三条或四条单链带就不足为奇。
为了高灵敏特异性地显示SSCP分析结果,现已发展多种PCR-SSCP技术,各有其优势及适用领域。例如,可以在PCR扩增中用同位素或荧光素等标记引物或核苷,也可以在电泳后用银染或溴化乙锭染色以显示结果。这里将重点介绍最常用的放射性同位素PCR-SSCP法。在进行PCR扩增特定靶基因序列时,利用γ-32P-ATP标记引物或直接在PCR反应体系中加入α-32P-dCTP进行PCR扩增,使扩增产物带有同位素标记物,然后将扩增产物变性为单链进行非变性聚丙烯酰胺凝胶电泳,放射自显影显示结果。利用引物标记或碱基掺入法使PCR扩增产物带有同位素标记物,均可使产物信号增强几个数量级。二者相比较,前者经济、扩增特异性强,多用于大样本的检测和筛选;后者操作简便,适于一般实验室开展,可用于小样本或大样本的检测和筛查。本节仅介绍同位素标记碱基掺入法。
一、试剂准备
⒈ PCR相关试剂
⒉ α-32P-dCTP
⒊ 30%聚丙烯酰胺(29:1):丙烯酰胺29g、甲叉双丙烯酰胺 1g,溶于100ml ddH2O中,4 OC保存。
⒋ 10%过硫酸胺配制方法:1g 过硫酸胺,溶于10ml ddH2O中,4 OC保存(可用数周)。
⒌ TEMED(N,N,N,N’,N’-四甲基乙二胺)
⒍ 5×TBE缓冲液:Tris碱54g、硼酸27.5g、0.5M EDTA(pH 8.0) 20ml,加ddH2O至1000ml。
7.变性上样液:95% 甲酰胺、0.03%二甲苯青、0.05%溴酚蓝、 20mM EDTA(pH 8.0)
二、操作步骤
⒈ PCR扩增
反应总体积为10μl,在0.5ml微量离心管中加入下列反应成分:
10×buffer 1μl
dNTPmix 70μM
DNA模板 100ng
引物及Taq DNA酶 依实验设计要求按比例加入
α-32P-dCTP 0.1μl
加ddH2O至 10μl
按实验设计循环参数进行扩增,获取扩增产物。
⒉ 聚丙烯酰胺凝胶电泳
⑴ 电泳槽玻璃板的处理:用洗涤剂清洗玻璃板,自来水反复冲净洗涤剂,双蒸水冲洗3次,晾干,95%乙醇擦拭,自然干燥。用棉签沾取SigmaCote涂于玻璃的贴胶面上, 5~10min后再用软纸擦拭除去多余的SigmaCote溶液。在两块玻璃内的两侧放好衬条对齐,用固定夹夹紧两块玻璃,并用玻璃胶带封边。
⑵ 制胶:按照被分离DNA片段的大小、含量及玻璃板、衬条的大小决定凝胶的浓度与体积,一般来讲使用5%~8%的凝胶较为合适。轻轻摇匀配制的胶液于真空抽气(开始时要缓慢)除气泡,加 35μl TEMED至聚丙烯酰胺混合液中,混匀。用10ml玻璃吸管或50ml注射器吸取胶液,将玻璃模具倾斜成60O角,缓慢注入两玻璃板间的空隙中,直至灌满模具顶部。立即插入相应的点样梳,(小心勿使梳齿下带进气泡,并且不要将梳齿全部插入胶内,留约2mm梳齿于玻璃板上端,以免拔梳时把胶孔拔断)。由于凝胶在聚合过程中有回缩,所以应小心添加些胶液于梳子处,水平放置,室温聚合1hr。将封口玻璃胶带掀去,放入电泳槽,凹型玻璃贴紧电泳缓冲液槽,用大号固定夹固定住两侧。在上下电泳槽内灌入 1×TBE电泳缓冲液。小心取出点样梳,用槽内的缓冲液反复冲洗点样孔,以去除可能存在的未聚合的聚丙烯酸胺和气泡。
⑶ 扩增产物的处理:将 PCR扩增产物与变性上样液按1:5的比例加入0.5ml 微量离心管中,混匀。样品上胶前应98 o C变性10min,立即冰浴骤冷。取约3~5μl变性样品(根据点样孔的大小决定上样量),以微量加样器上样,(上样时要注意不要有气泡冲散样品,而且速度要快,时间长了样品易于扩散)。
⑷ 电泳:电泳槽接上电极(上槽接负极,下槽接正极)开启电源,根据扩增片段的大小及电泳槽和凝胶的大小,决定电泳的电压及电泳时间。通常室温下以l~5V/cm电泳。
⑸ 剥胶:电泳结束后,倒弃电泳缓冲液,取下电泳胶玻璃,用塑料楔子从玻璃板底部一角小心分开玻璃,凝胶应附着在一块玻璃上,切去凝胶左上角,作为点样顺序标记。剪一张与玻璃同样大小的滤纸,严密覆盖于凝胶上,顺一个方向将凝胶缓慢取下,用保鲜膜盖于胶面并包好,避免膜和胶之间产生气泡或皱折,将凝胶固定在X线片夹中。
⑹放射自显影:在暗室中将X线片贴于胶面,盖严片夹,用黑布将X线片夹包裹,置-70OC放射自显影。曝光时间根据同位素的强度而定,约 24h~10d。
⑺ 冲洗胶片从-70oC取出X线片夹,在暗室内迅速取出X线片,立即显影,以免使X线片上出现过多的冷凝水。(如果想再得到一张放射自显影影像,可立即在片夹中装一张新X线片并尽快放回到一70oC继续显影。如果来不及再加入新片即已出现冷凝水则应使样品凝胶和X线片夹都恢复到室温,并擦去冷凝水后再装入新的X线片)。依次按以下程序操作进行显影:
X线片显影液显影 1-5min
水洗 lmin
定影液定影 5min
流动水冲洗 15min
所有使用液的温度应为18~20OC为宜。
三、注意事项
⒈ 核酸片段的大小: 用于SSCP分析的核酸片段越小,检测的敏感性越高。对于<200bp的片段,SSCP可发现其中70%的变异;对于300bP左右的片段则只能发现其中50%的变异;而>500bp的片段,则仅能检出10%~3O%的变异,因此,<300bP,尤其是150bP左右的核酸片段更适于SSCP分析。对于大于400bp的PCR产物就需要设法进一步处理,可以用限制性酶消化PCR产物,产生小于400bP的DNA片段,再进行SSCP分析。
⒉ 游离引物: 游离引物可能同 PCR产物结合而改变其泳动率,即使游离引物量为6nM都有明显影响。因此,应尽可能除去游离引物。可以采用不对称引物扩增方法,尽可能消耗多余的引物。也可以运用过柱或磁性球方法纯化PCR产物。或者是稀释PCR产物,减少游离引物的干扰。
⒊ 低浓度变性剂: 凝胶中加入低浓度的变性剂,如5%-10%甘油、5%尿素或甲酸胺、10%二甲亚砜(DMSO)或蔗糖等有助于提高敏感性,可能是因为轻微改变单链DNA的构象,增加分子的表面积,降低单链DNA的泳动率。但有些变异序列却只能在没有甘油的凝胶中被检出。因此,对同一序列使用2~3种条件做SSCP,可能提高敏感性。
⒋ 电泳温度: 一般认为保持凝胶内温度恒定是SSCP分析最关键的因素,温度有可能直接影响DNA分子内部稳定力的形成及其所决定的单链构象,从而影响突变的检出。室温下电泳适于大多数情况,但由于在电泳时温度会升高,为确保电泳温度相对恒定,应采取以下措施:减少凝胶厚度,降低电压,有效的空气冷却或循环水冷却等。
⒌ 凝胶的长度: 可用测序板进行SSCP分析,凝胶板长度在40cm以上。
⒍ 凝胶浓度及厚度: 凝胶浓度很重要,一般使用5%~8%的凝胶,凝胶浓度不同,突变带的相对位置也不相同,如果在进行未知突变种类的SSCP分析时,最好采用两种以上凝胶浓度,这样可以提高突变种类的检出率。凝胶的厚度对SSCP分析也很重要,凝胶越厚,背景越深,在上样量较多的前提下,尽量使凝胶越薄越好。
⒎ 假阴性: 一般认为,如没有污染,PCR-SSCP分析不存在假阳性结果,但可能出现假阴性结果,后者是由于点突变引起的空间构象变化甚微,迁移率相差无几所致,尤其是点突变发生在扩增片段的两端时。如果有阳性和阴性对照,结果可以重复确定的突变带是可信的,如果没有阳性对照,应经测序来确定其是否为突变带。由于PCR-SSCP的不足之处主要是可能检出假阴性结果。应通过设置阳性对照,摸索电泳条件,假阴性结果在很大程度上是可以避免的。但对未知基因变异的检测,假阴性结果就难以百分之百地消除。
8. 结果分析: 单链凝胶电泳时,互补单链迁移率不同,一般形成两条单链带。PCR产物进行单链凝胶电泳之前,通过加热变性产生单链。如变性不彻底,残留双链亦可形成一条带.因此,PCR-SSCP分析结果至少显示三条带。但是,由于一种DNA单链有时可形成两种或多种构象,检出三条或四条单链带就不足为奇。