RNA抽提- 成功经验法则大公开
互联网

本期专文要来跟大家讨论的话题是核糖核酸(RNA)的萃取,看到这里各位心中可能已经起了疑问:RNA 萃取不是很简单吗?只要照着标准步骤操作,加一加试剂就完成了,有必要大费周章特地开篇幅来谈这件事吗?其实越简单的东西遇到困难时就越难解,各位看倌就请耐着性子,听我们娓娓道来。
Lab 的经验和客户的难题
(1)RNA的总量不够
(2)RNA有严重的盐类污染
(3)RNA有严重的有机溶剂污染
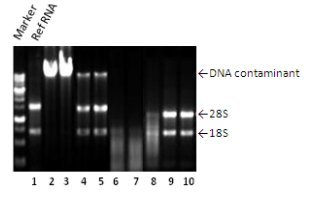
(4)DNA污染
(5)样品降解严重。
前四项问题都还好解决,只需要再次萃取或进行纯化即可解决,但是样品降解就较为棘手,常常因重新准备检体一来一往,耗掉不少心力和时间,最后也拿不到好的结果。
客户经常会问为什么执行芯片实验会如此要求RNA质量呢?而其他的检测方法如实时定量聚合连锁反应(Real-time Quantitative Polymerase Chain Reaction, Q-PCR) 却不需要?回答这个问题前就要从实验的原理说起,简单的讲Q-PCR的检测原理是针对要检测的基因,设计一对基因特异性的引子,透过反转录反应(RT)配合PCR的放大方式和及时侦测放大的产物量,藉以回推原始的RNA含量;而RT的方式主要是采用随机引子(Random_primer)、进行互补DNA (cDNA)的翻制,因此即便RNA有些微裂解,作为模板的RNA也可以忠实的被翻制成cDNA。
但是芯片实验进行,主要是以放大和讯息RNA(mRNA)完全配对的互补RNA(cRNA) ,再进行杂合反应,此步骤必须利用poly(dT)- T7启动子序列和讯息RNA的poly(A)结合后,将T7启动子序列连带的镶入反转录的cDNA的5端,再经由T7启动子驱动试管内反转录反应,生合成反股的cRNA,再进行后续的芯片杂交的实验。可想而知,如果RNA有降解严重的现象,表示许多mRNA并不能忠实的被放大因此会产生许多伪阴性的结果。
至此读者心中可能就有一个概念了,假设Q-PCR和芯片被应用在瞎子摸象的故事情节中,用Q-PCR的人只要能摸到大象的耳朵就可以说这里有大象,透过计算耳朵的数量就可以计算大象的数量;但是用芯片实验方法的人,必须先确认所摸到的个体具备大象的完整特征,才能确确实实的说这里有一只大象。这也就说明了为何芯片实验会如此要求RNA的质量。

RNA 萃取的方法有哪些?
除了传统的纯化方法,如 LiCl 沉淀等外,因应RNA研究的庞大需求,目前市面上已有许多琳琅满目的RNA萃取套组,供给不同萃取需求目的之研究,而这些产品的设计原理不外乎是利用有机溶剂进行分层萃取如:phenol-chloroform;或利用RNA于酒精中呈现疏水性的特性,进行吸附分离如:吸附性管柱 (Silica column)。一般而言,只要有正确的RNA操作观念配合正确的步骤,大致上都可以萃取出质量不错的RNA,但是不同的萃取方法所取得的RNA组成分布会有所不同,因此建议要根据所要分析的对象RNA,选择正确的萃取方法或套组,例如:如果要研究小RNA的表现就不可选择以mRNA为萃取对象的方法。此外,内源核醣核酸分解酶(RNase)含量高的样品,建议使用含phenol-chloroform的裂解液,以增加去除酶活性的能力。以下我就以TRIzol的操作步骤为例,来跟读者分享RNA萃取步骤中应注意事项及改善萃取RNA值与量的方法。
先前我们提及细胞内存在有内源性RNase,且其表现量在不同种类的组织中差异极大。例如脑组织和心脏组织拥有相对低的内源性RNase,但是胸腺组织和胰脏组织则保有约100,000倍于脑组织的内源RNase。因此动物组织等最好都先用液氮冷冻起来,再进行后续的处理,不建议直接丢到-80 ℃冰箱。因为急速冷冻可冻结RNase的活性,但-80 ℃冷冻算是缓慢降温,仍会造成部分RNA的降解;如果无法快速冷冻检体,也可以采用市售的检体保存液,亦可达到不错的效果。
有关样品的破碎和均质化,这个步骤的重点应该摆在如何让有机裂解液快速浸润检体,phenol-chloroform可以溶解细胞,连带的会造成蛋白变性,因此内源性RNase的活性就会去活化;相反的如果无法让细胞快速裂解使蛋白裸露进而变性,则RNase就会降解RNA,因此如何使不同组织细胞快速裂解,就是一个值得思考调整的课题,也是左右成败的关键。
就检体类型来说,一般培养细胞多为单层细胞不会遇到这样的问题,萃取较为容易,但是立体的组织进行萃取就会遇到均质化不均的问题进而造成RNA降解。因此低温的液氮碾磨就是破碎组织最有效的办法,但是液氮碾磨较为麻烦,如果遇到样品数比较多的时候耗时费功效力不彰,但如果条件允许的话,这仍然是一个好方法。但考虑效率和速度,均质机就是一个更好的选择,但是使用均质机的过程有几个重点必须注意:
(1)速度要快,在RNase展现活性前完成细胞均质化;(2)避免产热,以免降解 RNA;
(3)不要过度研磨,过度研磨会打断DNA,造成小片段的DNA污染。
此外,在均质裂解细胞的过程中有一个常被会忽略的重点,就是一般细胞和有机裂解液的比例常被忽略,有些操作者会认为多放一些组织就可以得到大量的RNA,但是没意识到太多的组织和太少的裂解液,造成细胞无法完全被裂解,RNase无法完全被变性,效果反而适得其反,最后RNA严重降解。因此如果反应完后均质液太过粘稠无法分层,就必须再加入裂解液。
在phenol-chloroform的萃取方式中,混合液的离心转数不可任意调整,应固定于12000 g的转数,以免因沉降系数改变造成无法有效分层。当离心完成后可以明显观察到检体分成上面水层及下面有机层,而我们要的RNA就溶解在上面水层中,体积约有500~600 μl左右。此时建议用小口径的 100 或 200 μl tip将水溶液分段吸取到干净的1.5 ml的离心管中,而在这个步骤常见的失误就是:
操作的人急于将水层取出而搅动原先的分层;或感觉萃取不易想要多回收一些RNA,于是将水层尽其可能的取出,因而造成吸取到有机溶液和大量的DNA污染,造成后续实验处理上的困扰甚至实验失败,因此强烈建议只萃取7~8成的RNA水溶液进行下一个步骤,俗话说的好,所谓有失必有得,不必为了一颗树而放弃整片森林,适时的取舍可以确保后续实验顺利进行。此外一般来说,phenol与水有一定比例的互溶,所以此时的RNA水溶液中仍有一定的phenol残留,建议一定要再使用chloroform进行一次离心分层去除残存phenol。

RNA 样品的沉淀
一般我们会使用乙醇(水溶液:乙醇=1:2.5)或异丙醇(水溶液:异丙醇=1:1)进行RNA沉淀,在效果上并无明显差异,但是如果已知RNA的含量是少的,则可以使用乙醇沉淀法,并将检体置于-80 ℃冰箱间隔一个晚上再做离心的动作,并配合离心时间的增加,如此可增加RNA沉淀的回收率。
为了去除RNA沉淀物中的盐类,我们可以采用70~75%乙醇进行洗涤,可能的话尽量让核酸沉淀悬浮起来,最好的是使用两次洗涤,再将离心管放入离心机中离心,用移液器将残留的乙醇小心吸掉,稍微抽干或吹干核酸沉淀物使残存的乙醇挥发后,再以DEPC水进行回溶。