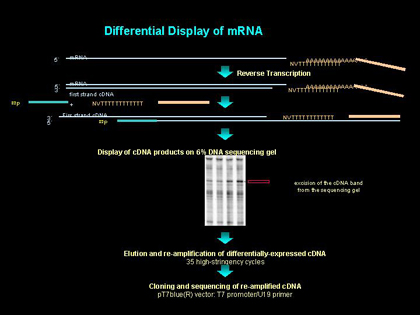
荧光标记mRNA差异显示技术
互联网
mRNA差异显示技术(differential display,DD)是用于研究基因的差异表达的新方法。该技术自1992年被首次报道后,即以其不可替代的优势被广泛应用于生物医学领域。在应用过程中不断得到改进,并产生了诸多衍生技术如RPA(RNA finger printing by arbitrarily primed PCR)、GDD(genomic DD)等。本文简要介绍在本试验室荧光标记差异显示技术(fluorescent DD,FDD)的应用及体会。
1.材料与方法
1.1 标本
人单核细胞系U937,细胞密度2×108/L,分对照组(N)、处理Ⅰ组(T1)、处理Ⅱ组(T 2),N用1640培养基及10%胎牛血清培养,T1用IFN-γ104U/L LPS 1μg/L、T2用IFN- γ10 U/L LPS l0μg/L分别刺激7h.
1.2 主要试剂与仪器
TRIzol试剂(GIBCO BRL)、Fluoro DD试剂盒(Genomyx)、Supersript Ⅱ逆转录酶(GIBCO BRL)、Ampli Taq DNA聚合酶(GIBCO BRL)、RNase-free DNaseI(Promega);Genomyx LR 、 Genomyx SC、DNAThermal Cycler(Perkin Elmer)
1.3 总RNA的制备
按试剂盒提供的方法分别提取三种细胞的总 RNA,以 RNase-free DNase I(终浓度80 000 U/L)除去其中污染的 DNA,经甲醛变性凝胶电冰鉴定其完整性,并以紫外分光光度计检 测其纯度
1.4 mRNA差异显示
1.4.1 逆转录反应
选择锚定引物[T7(dT12)AP(anchored prime rs,AP)],序列为5'ACGACTCACTATAGGGCTTTTTTTTTTTTMN3',其中 M=A/G/C。N=A /G/C/T],以总RNA为模板进行逆转录反应,每管反应体系如下:总RNA l.0μg,AP 4 pmol ,70℃ 5 min,加入50 mmol/L Tris-HCl(pH8.3),75 mmol/L KCl,3 mmol/L MgCl2,10mmol /L DTT,25μmol/L,dNTPmix(1∶1∶1∶1),SuperScriptⅡ 60 Units,总反应体系20 μl ,42℃ 5 min,50℃ 50 min,70℃ 15 min。
1.4.2 荧光标记差异显示PCR
选取与逆转录引物序列相同的带荧光物 质标记的锚定引物[TMR-T7(dT12)AP],随机引物(5'ACAATTTCACACAGGAACGCTAGTTG 3'),以逆转录产物为模板,进行PCR反应。反应体系包括:20 mmol/L Tris-HCl(pH8.4),50 mmol/L KCl,3.75 mmol/L MgCl2,逆转录产物3.0 μl,50 μ mol/L dNTP mi x(1∶1∶1),0.35 μmol/L 5'-随机引物,0.35 μmol/L 3-锚定引物,Ampli Taq 0.5 U nits,总反应体系10 μl。反应步骤如下:95℃ 2 min;94℃ 15 s,50℃ 30 s,72℃ 2 min,4个循环; 94℃ 15 s,60℃ 30 s,72℃ 2 min,个循环;72℃延伸7 min。
1.4.3 分离差异显示片段
配制5.6%变性聚丙烯酰胺凝胶,胶厚0.2 5 mm,大小61×33 cm。将PCR产物加4.0μl上样缓冲液,95℃变性后上样。3000V、100W 、55℃电泳4.5 h。干胶后置于Genomyx SC扫描,用系统所带的AcquireSC program软件分 析处理扫描结果。
1.4.4 回收差异条带
用AcquireSC软件将差异条带定位,用一次性手术刀片切割下所需条带,置于30μl去离子水中,37℃水浴30-60 min,备用。
1.4.5 差异条带的再扩增
以经上述处理的回收条带为模板,T7启动子22-mer(5'GTAATACGACTCACTATAGGGC3’)、反M13(-48)24-mer(5'AGCGGATAACAATTTCACACA GGA3')为引物,进行再扩增反应:模板2.0μl,20 mmol/L Tris-HCl(pH8.4),50 mmol/L KCl,1.5 mmol/L MgCl2,20 μmol/L dNTP mix(1∶1∶1∶1),0.2 μmol/L T7、0 .2 μmol/L M13-r、AmpliTaq 1.0 U nits,总反应体系20 μl.反应条件同差异显示PCR。
2.结果
2.1 总RNA的质量
总RNA经Dnase I处理,用1.0%甲醛变性凝胶电泳,可见清楚的28 S、18 S、5 S条带,且28 S/18 S约为2∶1(图1),表明RNA完整性好,无降解现象。N、T1、T2的OD260/OD28 0比值分别为1.92、1.83、1.98,表明样品纯度高。
Fig.1 Result of total RNA on formaldehyde-agarosegel
2.2 荧光标记mRNA差异显示
结果显示每一泳道均有约50条大小不等的扩增产物,各组间比较见较多呈有无或强弱变 化的差异条带,图2中箭头所示。
Fig.2 Analysis of gene expression of cells treated with IFN and LPS by differential display technique
2.3 差异条带再扩增
取T2中约1.25Kb的条带再扩增,结果见图3.
Fig.3 Reamplification result of differ entially expressed
band DNA marker is 200 bp ladder (200 bp~2 kb),1.0kb is arro wed
3. 讨论
基因表达是调节细胞生物学行为的核心。探讨处于不同生理、病理状态下的有机体之间 的未知表达基因的差异,是研究生命本质的有效途径。1992年报道的真核细胞mRNA差异显示技术,为检测未知的表达基因提供了一种新的途径。DD技术具有快速、敏感、 可同时检测两组或以上的组织或细胞、RNA用量少、可检测某一表达基因的有无或其表达的强弱等优点,一经问世即受到许多领域的重视并得以广泛应用。然而,该技术也存在着一些缺陷,主要表现为:cDNA产物的质量较低,在序列胶中往往呈不清晰状态;所得的cDNA片段通常小于500nt,往往是3'-端的非翻译(编码)序列;所得差异片段的假阳性高,可高达85%等。
荧光标记差异显示技术通过对引物设计、PCR条件、凝胶、电泳条件以及标记物的改进,极大减少了上述不足。
差异显示的锚定引物有单碱基锚定引物、简并或非简并的双碱基锚定引物等多种类型,本实验通过使用双碱基锚定引物,将总mRNA群体分为12个亚群,这极大减小了每一锚定引物 产生的第一条cDNA链的数量,不仅可降低随机引物的用量,更可降低DD产物的复杂性,使差示条带在序列胶上易于分辨,从而传统方法中常见的“重叠带”现象减少。DD专用的随机引物的特点为A+T与G+C含量近乎相等,且3'-端以G或C结尾,可增加DD扩增的效率。通过改变RT-PCR反应条件如底物浓度、延伸时间等,差异片段的长度可达1.0-1.5 kb(图2), 因较长的cDNA片段容易通过Northern blot 进行分析鉴定辨别真假阳性克隆,且其中可提供更多的序列信息,故获取大的片段具有重要的意义。
文献认为差异显示居高不下的假阳性率实际上因再扩增所致。通常, 再扩增的引物与DD引物相同,本试验将再扩增引物设计为T7(22-mer)与M13-r(24-mer),此为在锚定引物的5' 端和随机引物的3' 端分别增加的序列(本文方法中所列差异显示引物序列的 划线部分),这两种来源于原核生物的引物尽可能降低了在真核mRNA序列中非特异性扩增的机会。同时,T7启动子序列可直接用于体外转录合成cRNA探针,为cDNA片段的直接测序和鉴定(RPA或Northern分析)提供了方便。
用常规的序列分析胶进行分辨时,较长的cDNA片段往往挤在胶的上端而影响分辨率。本 操作改用5.6%的PAGE胶(聚丙烯酰胺),减少胶的厚度(仅250μm),通过提高电泳的电压(3000 V)及温度(55℃),可显着提高分辨率。
同位素标记存在曝光时间长、带型不佳,基本与相邻的带混合、污染等缺点,将荧光物 质标记于锚定引物的5’端,可产生清晰、明亮、低背景的分辨效果,操作周期仅两天。
目前,DD技术己被广泛应用于与疾病、衰老、生殖、生长发育、分化等生理 、病理过程相关的未知表达基因的研究,相信对揭示生物界基因表达调控的奥秘将起重要的作用。
1.4.5 差异条带的再扩增
以经上述处理的回收条带为模板,T7启动子22-mer(5'GTAATACGACTCACTATAGGGC3’)、反M13(-48)24-mer(5'AGCGGATAACAATTTCACACA GGA3')为引物,进行再扩增反应:模板2.0μl,20 mmol/L Tris-HCl(pH8.4),50 mmol/L KCl,1.5 mmol/L MgCl2,20 μmol/L dNTP mix(1∶1∶1∶1),0.2 μmol/L T7、0 .2 μmol/L M13-r,AmpliTaq 1.0 Units,总反应体系20μl。反应条件同差异显示PCR。
2. 结果
2.1 总RNA的质量
总RNA经Dnase Ⅰ处理,用1.0%甲醛变性凝胶电泳,可见清楚的28 S、18 S、5 S条带 ,且28 S/18 S约为2∶1(图1),表明RNA完整性好,无降解现象。N、T1、T2的OD260/OD280比值分别为1.92、1.83、1.98,表明样品纯度高。

Fig.1 Result of total RNA on formaldehyde-agarosegel
2.2 荧光标记mRNA差异显示
结果显示每一泳道均有约50条大小不等的扩增产物,各组间比较见较多呈有无或强弱变化的差异条带,图2中箭头所示。

Fig.2 Analysis of gene expression of cells treated with IFN and LPS by differential display technique
2.3 差异条带再扩增
取T2中约1.25Kb的条带再扩增,结果见图3。

Fig.3 Reamplification result of differ entially expressedband DNA marker is 200 bp ladder (200 bp~2 kb),1.0kb is arro wed
3. 讨论
基因表达是调节细胞生物学行为的核心。探讨处于不同生理、病理状态下的有机体之间 的未知表达基因的差异,是研究生命本质的有效途径。1992年报道的真核细胞mRNA差异显示技术,为检测未知的表达基因提供了一种新的途径。DD技术具有快速、敏感、 可同时检测两组或以上的组织或细胞、RNA用量少、可检测某一表达基因的有无或其表达的 强弱等优点,一经问世即受到许多领域的重视并得以广泛应用。然而,该技术也存在着一些 缺陷,主要表现为:cDNA产物的质量较低,在序列胶中往往呈不清晰状态;所得的cDNA片段通常小于500nt,往往是3'-端的非翻译(编码)序列;所得差异片段的假阳性高,可高达85%等。
荧光标记差异显示技术通过对引物设计、PCR条件、凝胶、电泳条件以及标记物的改进,极大减少了上述不足。
差异显示的锚定引物有单碱基锚定引物、简并或非简并的双碱基锚定引物等多种类型,本实验通过使用双碱基锚定引物,将总mRNA群体分为12个亚群,这极大减小了每一锚定引物产生的第一条cDNA链的数量,不仅可降低随机引物的用量,更可降低DD产物的复杂性,使差示条带在序列胶上易于分辨,从而传统方法中常见的“重叠带”现象减少。DD专用的随机引物的特点为A+T与G+C含量近乎相等,且3' 端以G或C结尾,可增加DD扩增的效率。通过改变RT-PCR反应条件如底物浓度、延伸时间等,差异片段的长度可达1.0-1.5 kb(图2),因较长的cDNA片段容易通过Northern blot 进行分析鉴定辨别真假阳性克隆,且其中可提供更多的序列信息,故获取大的片段具有重要的意义。
文献认为差异显示居高不下的假阳性率实际上因再扩增所致。通常, 再扩增的引物与DD引物相同,本试验将再扩增引物设计为T7(22-mer)与M13-r(24-mer),此为在锚定引物的5' 端和随机引物的3' 端分别增加的序列,这两种来源于原核生物的引物尽可能降低了在真核mRNA序列中非特异性扩增的机会。同时,T7启动子序列可直接用于体外转录合成cRNA探针,为cDNA片段的直接测序和鉴定(RPA或Northern分析)提供了方便。
用常规的序列分析胶进行分辨时,较长的cDNA片段往往挤在胶的上端而影响分辨率。本操作改用5.6%的PAGE胶(聚丙烯酰胺),减少胶的厚度(仅250μm),通过提高电泳的电压(3000 V)及温度(55℃),可显着提高分辨率。
同位素标记存在曝光时间长、带型不佳,基本与相邻的带混合、污染等缺点,将荧光物质标记于锚定引物的5' 端,可产生清晰、明亮、低背景的分辨效果,操作周期仅两天。
目前,DD技术己被广泛应用于与疾病、衰老、生殖、生长发育、分化等生理 、病理过程相关的未知表达基因的研究,相信对揭示生物界基因表达调控的奥秘将起重要的作用。