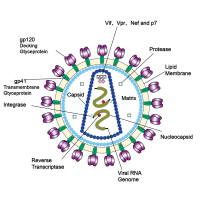
载体质粒的抽提、纯化及检测
互联网
一、目的:
了解质粒抽提常用的方法和原理;掌握试剂盒制备质粒DNA 的法。
二、原理:
(一)质粒DNA 制备三个步骤:
1、细菌培养与质粒扩增
2、菌体收集和溶菌
3、质粒DNA分离
(二)方法介绍:
1、碱变性抽提法(SDS法):主要是利用染色体DNA和质粒DNA碱性环境中变性和复性的差异来分离出质粒DNA。染色体DNA 分子量比较大,在碱性环境中(高pH)变性,双链解拆开,抽提时由于机械剪切力的作用,环状断裂成线状,当pH 恢复中性时,无法恢复成环状,就呈网状与蛋白质一起通过离心沉淀下来。
质粒DNA 分子量小,在碱性环境中(高pH),双链解旋,但不拆开,当pH恢复中性时,恢复成环状。由于分子量小,所以离心沉淀时,不下沉,保留在上清溶液中。
缺点:SDS 破细胞很厉害,小染色体片段和蛋白质比较多,后处理较困难。
2、酸酚法:主要是利用染色体DNA和质粒DNA 在酸性环境中变性和复性的差异来分离出质粒DNA。
染色体DNA 分子量比较大,在酸性环境中(低pH)变性,双链解拆开,抽提时由于机械剪切力的作用,环状断裂成线状,当pH 恢复中性时,无法恢复成环状,就呈网状与蛋白质一起通过离心沉淀下来。
质粒DNA 分子量小,在酸性环境中(低pH),双链解旋,但不拆开,当pH恢复中性时,恢复成环状。由于分子量小,所以离心沉淀时,不下沉,保留在上清溶液中。
缺点: pH 值太低会降解DNA, pH 的适度难以掌握,稍一偏差,全部DNA 被降解,什么也没抽到。现不常用。
3、溴化乙锭-氯化铯(CsCl)梯度离心法:
1)不同的氯化铯浓度和不同的离心时间会成为不同的密度梯度。
2)抽提破细胞后,蛋白质,染色体DNA,质粒DNA,RNA 全部释放出来,加入溴化乙锭(EB),通过EB对不同物质的插入多少而形成不同的密度梯度。EB插入越多,密度越小。离心后,在离心管中从上到下(密度由小到大)形成的梯度区域带如下:
↓
蛋白质(密度最小)
染色体DNA(机械剪切力的作用断裂成线状,EB插入较多,密度较小)
质粒DNA-线状(EB 插入较多,密度较小)
质粒DNA-环状(EB 插入较少,密度较大)
质粒DNA-超螺旋(EB 插入少,密度大)
RNA(单链,EB 插入最少,密度最大)
↑
不同构象的核酸(线形、环形、超螺旋),密度和沉降速率不同,用Cs-Cl 密度梯度离心可以将不同构象DNA、RNA 与蛋白质区分开来
优点:此法抽提的质粒DNA 高纯度。
缺点:1)要求高档的超速离心机。
2)成本高,氯化铯价格贵。
4.TritonX-100 抽提法:
TritonX-100为非离子极性剂,在溶液中不形成离子,破细胞比较温和。使用该试剂是为了破细胞时不要太激烈,只形成孔状破裂,使质粒DNA 释出(当然还有RNA和一些小片段的染色体DNA 和蛋白质),而不释出大片段的染色体DNA 。当离心时染色体DNA的大片段与细胞膜的网状碎片附在一起,与蛋白质一起被离心沉淀,而质粒DNA则留在上清。所以细胞膜破裂的程度是实验的关键。
然后使用苯酚与氯仿来祛除蛋白质。用无水乙醇沉淀回收质DNA。
优点:抽提的质粒比较纯,较好用, TritonX-100 不影响后续实验,可以大量抽提。
缺点:比试剂盒抽提时间要长
三、试剂盒抽提法(本实验采用):
(一)抽提原理:
人们使用碱与SDS裂解法从.E.coli 中分离制备质粒DNA 已经有20 多年的历史。将细菌悬浮液暴露在高pH值的强阴离子洗涤剂(如NaOH和SDS)中,会使细胞壁破裂,染色体DNA 和蛋白质变性,将质粒DNA释放到上清中。尽管碱性溶剂使碱基配对完全破坏,闭环的质粒DNA 双链仍不会彼此分离,这因为他们在拓扑学上是相互缠绕的(超螺环结构)。只要碱处理的强度和时间不要太过,当pH 值恢复到中性时,DNA 双链就会再次形成。在裂解过程中,细菌蛋白质、破裂的细胞壁和变性的染色体DNA会相互缠绕成大型复合物,后者被SDS 包盖。当用钾离子取代钠离子时,这些复合物会从溶液中有效地沉淀下来。
离心除去沉淀,就可以从上清中回收复性的质粒DNA。
早在20 世纪50 年代,人们就已经知道,在较低pH 值和高盐浓度环境下,DNA 能与硅胶(Silica gel)可逆结合。DNA双链与硅化材料相互作用的原理普遍认为是DNA分子中磷酸二酯键在较低pH 值和高盐浓度环境下脱水,使得暴露的磷酸基团吸附硅胶。双链的DNA分子一旦被硅胶吸附,则以天然状态或部分变性状态存在,不能用洗脱RNA或碳水化合物等生物大分子的溶剂(如70%乙醇)将其洗脱下来。但是,经过水溶性缓冲液(通常为TE或水)重新水化后,DNA 就能从层析柱上定量回收。
优点:快且纯。
(二)所用材料、试剂与器材:
1.高速离心机、微量移液器、1.5ml EP管
2.Rapid Plasmid DNA Daily Mini-prep Kit:
3.DNA-prep Tube(Silica 膜)
4.2ml Microfuge Tube
5.Buffer S1(葡萄糖维持渗透压、Tris-HCl 维持pH 值、EDTA 抑制核酸酶、RNaseA1降解RNA)
6.Buffer S2(NaOH裂解细胞与核酸变性、SDS裂解细胞与蛋白质变性)
7.Buffer S3(乙酸钾置换钠离子、冰乙酸调节pH 值)
8.BufferW1(乙酸钠洗脱蛋白质、糖类等大分子杂质)
9.BufferW2(70%乙醇洗脱盐离子)
10.Eluent(TE pH8.5 洗脱质粒DNA)
11.大肠杆菌C600(内含pUC18 质粒)
12. LB 完全培养基液(1%蛋白胨 0.5%酵母粉 0.5%NaCl pH7.5。)
(三)操作步骤与注意事项:
1.用接种棒取一环保存的大肠杆菌C600(内含pUC18 质粒)在LB 完全培养基斜面上接种,活化菌株。
2.在5 毫升LB培养液中,用微量进样器加入5微升氨苄青霉素溶液。同时接入已活化的大肠杆菌C600 一环,37℃援床培养过夜。
3.取100 微升氨苄青霉素溶液于100 毫升LB 培养液中,然后取1 毫升过夜培养物转接进去(1%接种量),37℃摇床培养过夜。
4.第二天分组用1.5ml EP 管收集细胞:将菌液10,000 rpm 离心1m,弃尽上清,重复一次,将管口倒置在纸巾上,轻轻敲击,使上清流出,管壁上不应有残留上清。
5.加入250μl BufferS1,涡旋,充分悬浮细菌沉淀。悬浮需充分,对光观察,不应留有小的菌块,否则会影响菌体的裂解。
6.加入250μl BufferS2,温和但充分地上下翻转4-6 次,此步骤不宜超过5m。避免剧烈混合,否则将导致基因组DNA 的污染。
7.加入400μl BufferS3,温和地上下翻转10-20 次,直至沉淀由疏松状态变为相对紧密,体积明显缩小为止,室温静置2m。12,000rpm 离心10m。避免剧烈混合,否则将导致基因组DNA 的污染。若离心后凝结块未沉淀沉淀到管底部,应再次翻转混合数次,12,000rpm离心3m。