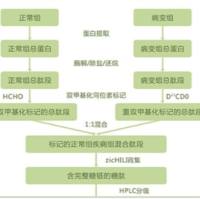
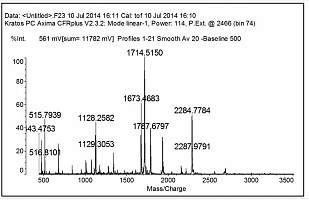
利用纳升级液相色谱- 串联质谱进行蛋白质鉴定
丁香园
4977
1. 前言
蛋白质组学分析是一个多步骤的过程,通常涉及蛋白质的提取、分级、分离和质谱鉴定。分离蛋白质的首选是二维电泳。胶内消化、纯化的蛋白可以利用 MS 或 MS/MS ( 串联质谱)得到的肽段进行分析、鉴定。如果已知物种的基因全序列,则通常使用基质辅助激光解吸质谱法电离飞行时间(MALDI- TOF) 的 MS 光谱技术来进行分析。它是一种高通量鉴定蛋白质的方法,而且也不需要纯化肽段。另外,尽管 MS/MS 分析比较费时,但可以在没有数据库(从头测序)情况下识别单个肽段。理论上,利用 MS/MS 鉴定出的一个肽段就可足以找到相应的蛋白质,而实际上利用 MS 鉴定蛋白则至少需要 4 个或 5 个肽段和肽质量指纹图谱(peptide mass fingerprinting) 。在只有不完整的序列数据库 [ 如只有表达序列标签(EST ) 数据库 ] 或只有其他物种的保守序列用于识别时,MS/MS 具有较大的便利性。与二维电泳相比,提出了另外一种简单水解蛋白质混合物的方法而不需要分离蛋白质:利用多维液相色谱(LC ) 和 MS/MS 用于分离复杂混合物和识别数千条多肽片段。这两种方法之间,中间的方法是使用十二烷基硫酸钠聚丙烯酰胺(SDS-PAGE ) 凝胶电泳分离部分蛋白质和利用 LC-MS/MS 进行蛋白质鉴定 [ 4~6 ] 。
蛋白质组学的 MS/MS 主要采用配备一个电喷雾源的离子阱(IT ) 和四极杆飞行时间(Q-TOF) 仪器。电喷雾是利用质谱仪的金属喷针(needle) 和入口之间的电位差而产生的。这会导致喷针出口处的液滴带电。液滴蒸发成细小的喷射流后,质谱仪根据质荷比(m/Z) 可以检测离子(带电肽段)。在 Q-TOF 质谱仪中,第一个样品池将电喷雾源的离子转移到四级杆中。四级杆设置传输一个离子(前体离子)到碰撞池。在碰撞池中,前体离子进行裂解。裂解碎片(裂解离子)在另一个质量分析器(TOF 管)中进行分析。由于碎片是沿着肽段骨架裂解的,因此氨基酸序列可根据碎片离子扫描(MS/MS 扫描)进行测定。在离子阱 (IT ) 质谱仪中,离子的选择、片段裂解和片段分析都是在同一池子(离子阱)进行的。实际上,MS/MS 仪器可以进行 MS 和 MS/MS 之间的切换。当离子选择和碎片裂解关闭时,MS 就可以进行扫描。MS 扫描可以检测所有的肽段离子和进行片段裂解。
电喷雾对无机盐非常敏感,因此肽段样品上样之前必须进行脱盐处理。最有效的一种方法是使用反相高效液相色谱法(RP- HPLC) 。反相高效液相色谱法不仅可以去除盐类 ,而且还可以纯化和浓缩肽段样品。这就是为什么液相色谱- MS/MS (LC-MS/MS)是蛋白质组学中最常见的配制。由于电喷雾技术跟样品浓度高度相关,色谱柱的直径是主要参数,减少柱子的直径可以提高灵敏度 [7] 。目前用直径 50~100 μm 的纳米级色谱柱来提高质谱仪进样口的肽段浓度。此外,利用纳米级色谱柱将流量减少到 150~300 nl/min 也可以提髙电喷雾的有效性。
在过去的十几年中,nanoLOMS/MS 技术已被广泛使用。在大多数质谱实验室,它已成为一种普遍的技术。由于电喷雾电离(ESI) 不同于 MALDI 电离过程,因此分析出的肽段结果有所不同。这些差异可以解释为什么 nanoLC- MS/MS 是 MALDI-TOF 质谱分析的一个有效补充 [ 9,10 ]。
2. 材料
2.1 胰蛋白酶消化
( 1 ) 天然聚丙烯聚合酶链反应(PCR ) 的 12 联管(0.2 ml/管)和相应的冷冻贮存盒架子(VWR International , Fontenay - sous - Bois, France)。
( 2 ) 多通道移液枪(YWR International ) 。
( 3 ) Eppendorf Repeater 分液器和 Combitips 分液管(VWR International ) 。
( 4 ) Eppendorf 热混合器 Thermom ixer ( Eppendorf, LePecq,France ) 和用于加热联管的模块加热块(VWR International ) 。
( 5 ) 修饰胰蛋白酶(Promega,Madison,USA ) 母液(200 ng/μl ) 。
( 6 ) 脱色液:10% 乙酸的 40% 甲醇(用 dH2O 配制)。
( 7 ) 清洗液:50 mmol/L NH4HCO3 ( 用 dH2O 配制,母液 1 mol/L ) 。
( 8 ) 消化缓冲液:25 mmol/L NH4HCO3,20% 甲醇(用 dH2O 配制)。
( 9 ) 收缩液:100% 乙腈。
( 10 ) 萃取液:0.5% 三氟乙酸(TFA ),40% 乙腈,10% 甲醇(用 dH2O 配制)。
2.2 高效液相色谱仪(NanoLC)
( 1 ) NanoHPLC 终极系统:Famos-Switchos-Ultimate ( Dionex-LC Packings,France) 。
( 2 ) C18 反相柱:300 μm X 5 mm ( trap column ) 和 75 μm X 10 cm ( analyticalcolumn ; PEPMAPC 18,Dionex LC- Packings)。
( 3 ) 流体管路接头(fitting) 和管路(tubing) ( Upchurch) 。
( 4 ) 熔融石英管(Upchurch) 。
( 5 ) 样品溶液:0.1% TFA,2% 乙腈(用 dH2O 配制)。
( 6 ) 装载溶剂(清洗捕获柱子,7 μl/min,见图 20-1): 0.1% 甲酸,0. 02% TFA,2% 乙腈。
( 7 ) 高效液相色谱溶剂A : 0.1% 甲酸,2% 乙腈(用 dH2O 配制)。
( 8 ) 高效液相色谱溶剂B : 用 95% 乙腈和 5% dH2O 配制的 0.1% 甲酸。
2.3 质谱仪(离子阱)
( 1 ) 纳米级喷针 360/20/10,未加涂层(New Objective,Woburn ) 。
( 2 ) 离子讲质谱仪,LCQ XP Plus ( Thermo Electron,Courtaboeuf,France) 。
( 3 ) 数据采集软件,Xcaliburl. 3 ( Thermo Electron) 。
( 4 ) 数据处理软件,BioWorks 3.1 ( Thermo Electron) 。

4. 注释
( 1 ) 建议以一系列代表性的样品在加或不加 DTT/IAA 情况下验证还原和烷基化反应的效率(或无效性)。检查已鉴定肽段的数量,角蛋白的污染,和是否存在羧胺甲基化半胱氨酸(+ 57 Da) 。
( 2 ) SpeedVac 会导致颗粒和干燥蛋白的溶解性减小,降低肽段的回收率。30 μl 的提取物在室温下需要过夜蒸发或在 4°C 蒸发 1~2 天。
( 3 ) 液体接头应紧固耐用,足以运行数百个样品而不会有任何问题(在堵塞的情况下除外)。
( 4 ) 液体接头的电极丝必须抗氧化。可以使用电泳仪用的销金线。
( 5 ) 分流是一种有效的方法,可以减少泵、泵管、混和器的死体积。另外,流速取决于来自分流管(nanoLC 系统的一般为 10 μm 内径)和柱子的压力。这些压力的任何变化都会改变流速。利用活动分流系统可以改善这一性能。
( 6 ) 在 m/z 大范围进行全质谱扫描是 “低分辨率” 扫描。在 m/z 短范围内(10 m/z 集中在父本离子)进行变焦 MS 扫描是 “高分辨率” 扫描。这种扫描可以测定肽段的电荷和质量。这里为 MS/MS 选择了两个离子。选定离子的数量取决于肽段的洗脱时间、肽段混合物的复杂性、每次扫描的速度以及仪器的灵敏度。对于复杂混合物,不使用 MS ZoomScans,增加选定离子的数量(最多 5 个) 。这就会增加待分析肽段的数量。
( 7 ) 纳流喷雾源安装摄像头,可以让用户看到喷头和喷雾,便于调整和故障排除 。当 LC 泵开始运行接通电压后,液滴必须喷出,喷头上不能有任何液体。
( 8 ) 在 http://www. abrf. org/ABRFNews/1996/Septemberl996/sep96iontrap . html 可找到有关 IT 和 Qz 对除去低质量的影响较为全面的教程。
( 9 ) 动态排除列表中对应的是父本离子的质量,这些是在最后几秒钟检测得到的。这可以防止对相同的父本离子进行重复 MS/MS 扫描。此外,这种设置还可以分析共洗脱时流出的低丰度的离子。
( 10 ) EST 是 DNA 序列(一般含 200~500 个核苷酸),是在系统地对 cDNA 文库的两端进行测序时所产生的。EST 重叠群是 EST 簇的共有序列。单一序列是跟任何 EST 没有相似性的 EST。EST 重叠群和 EST 单一序列通常分组;相应的数据库称作假定独立基因(tentative unique genes,TUG ) 。TUG = TUC + TUS。 TUC 和 TUS 分别是假定独立重叠群( tentative unique contigs) 和假定独立单一序列(tentative uniquesinglets) 的缩写 。
( 11 ) 参见 IPI ( 国际蛋白索引)ftp : //ftp . ebi. ac. uk/pub/databases/IPI/current/。新的标准有最新数据库:ftp ://ftp.ncbi. nih.gov/blast/db/FASTA / 和植物的蛋白序列数据库: http://www.plantgdb.org/download.p hp。
( 12 ) 选择修改变量会增加索引数据库的大小,延长查询时间并降低相关性得分。除了蛋氨酸氧化外,在第一轮查询时不必修改任何变量,以降低鉴定假象。
( 13 ) 在 MS/MS 分析中,角蛋白和胰蛋白酶肽段的少量污染不算什么问题。只有当它们的峰是最强的峰时才成为难题,在这种情况下感兴趣的肽段对应的小峰,可能会被屏蔽而不能分析。
参考文献
1. Washburn, M . P., Wolters, D., and Yates, J. R. 3rd. (2001) Large- scale analysis ofthe yeast proteome by multidimensional protein identification technology. Nat.Biotechnol. 1 9 , 242-247.
2. Froehlich, J. E., Wilkerson, C. G., Ray, W . K., et al. (2003) Proteomic study of theArabidopsis thalianachloroplastic envelope m e m b r a n e utilizing alternatives totraditional two-dimensional electrophoresis. J. Proteome Res.2, 4 1 3 -42 5 .
3. Lin, D., Tabb, D. L., Yates, J. R. Ill (2003) Large-scale protein identificationusing mass spectrometry. Bioch. Biophys. Acta 16 4 6, 1- 10.
4. Ferro, M., Salvi, D., Brugiere, S., et al. (2003) Proteomics of the chloroplast envelope m e m b r a n e s from Arabidopsis thaliana. Mol. Cell. Proteomics2, 325-345.
5. Carter, C., Pan, S., Zouhar, J., Avila, E. L., Girke, T., and Raikhel, N. V. (2004)T he vegetative vacuole proteome of Arabidopsis thalianareveals predicted andunexpected proteins. Plant Cell. 16, 3285 -3303.
6. Friso, G., Giacomelli, L., Ytterberg, A. J., et al. (2004) In-depth analysis of thethylakoid m e m b r a n e proteome of Arabidopsis thalianachloroplasts: n e w proteins,n e w functions, and a plastid proteome database. Plant Cell. 16, 478-499.
7. Abian, J., Oosterkamp, A. J., and GelpI, E. (1999) Comparison of conventional,narrow-bore and capillary liquid chromatography/mass spectrometry forelectrospray ionization mass spectrometry: practical considerations. J. MassSpec from.34, 244-254.
8. Wilm, M . and Man n , M . (1996) Analytical properties of the nanoelectrospray ionsource. Anal. Chem.68, 1-8.
9. Bodnar, W . M., Blackburn, R. K., Krise, J. M., and Moseley, M . A. (2003)Exploiting the complementary nature of L C / M A L D I / M S / M S and LC/ESI/MS/M S for increased proteome coverage. J. Am. Soc. Mass Spectrom. 1 4 , 971-979.
10. Lim, H., Eng, J., Yates, J. R. 3rd, et al. (2003) Identification of 2D-gel proteins: acomparison of M A L D I / T O F peptide mass mapping to m u L C - E S I tandem massspectrometry. J. Am. Soc. Mass Spectrom. 1 4 , 957-970.
11. Schagger, H. and von Jagow, G. (1991) Blue native electrophoresis for isolationof m e m b r a n e protein complexes in enzymatically active form. Anal. Biochem.1 99 , 223-231.
12. Davis, M . T. and Lee, T. D. (1997) Variable flow liquid chromatography-tandemmass spectrometry and the comprehensive analysis of complex protein digest mixtures. J. Am. Soc. Mass Spectrom.8, 1059-1069.
13. Schlosser, A. and Volkmer-Engert, R. (2003) Volatile polydimethylcyclosiloxanes in the ambient laboratory air identified as source of extreme backgroundsignals in nanoelectrospray mass spectrometry. J. Mass. Spectrom.38, 523-525.
14. Roepstorff, P. and Fohlman, J. (1984) Proposal for a c o m m o n nomenclature forsequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601.
15. Yates, J. R. 3rd, Eng, J. K., and M c C o r m a c k , A. L. (1995) Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequencesin nucleotide databases. Anal. Chem.67, 3202-3210.
16. Eng, J., M c C o r m a c k , A. L., and Yates, J. R. III. (1994) A n approach to correlatetandem mass spectral data of peptides with amino acid sequences in a proteindatabase. J. Am. Soc. Mass Spectrom.5, 976-989.
蛋白质组学分析是一个多步骤的过程,通常涉及蛋白质的提取、分级、分离和质谱鉴定。分离蛋白质的首选是二维电泳。胶内消化、纯化的蛋白可以利用 MS 或 MS/MS ( 串联质谱)得到的肽段进行分析、鉴定。如果已知物种的基因全序列,则通常使用基质辅助激光解吸质谱法电离飞行时间(MALDI- TOF) 的 MS 光谱技术来进行分析。它是一种高通量鉴定蛋白质的方法,而且也不需要纯化肽段。另外,尽管 MS/MS 分析比较费时,但可以在没有数据库(从头测序)情况下识别单个肽段。理论上,利用 MS/MS 鉴定出的一个肽段就可足以找到相应的蛋白质,而实际上利用 MS 鉴定蛋白则至少需要 4 个或 5 个肽段和肽质量指纹图谱(peptide mass fingerprinting) 。在只有不完整的序列数据库 [ 如只有表达序列标签(EST ) 数据库 ] 或只有其他物种的保守序列用于识别时,MS/MS 具有较大的便利性。与二维电泳相比,提出了另外一种简单水解蛋白质混合物的方法而不需要分离蛋白质:利用多维液相色谱(LC ) 和 MS/MS 用于分离复杂混合物和识别数千条多肽片段。这两种方法之间,中间的方法是使用十二烷基硫酸钠聚丙烯酰胺(SDS-PAGE ) 凝胶电泳分离部分蛋白质和利用 LC-MS/MS 进行蛋白质鉴定 [ 4~6 ] 。
蛋白质组学的 MS/MS 主要采用配备一个电喷雾源的离子阱(IT ) 和四极杆飞行时间(Q-TOF) 仪器。电喷雾是利用质谱仪的金属喷针(needle) 和入口之间的电位差而产生的。这会导致喷针出口处的液滴带电。液滴蒸发成细小的喷射流后,质谱仪根据质荷比(m/Z) 可以检测离子(带电肽段)。在 Q-TOF 质谱仪中,第一个样品池将电喷雾源的离子转移到四级杆中。四级杆设置传输一个离子(前体离子)到碰撞池。在碰撞池中,前体离子进行裂解。裂解碎片(裂解离子)在另一个质量分析器(TOF 管)中进行分析。由于碎片是沿着肽段骨架裂解的,因此氨基酸序列可根据碎片离子扫描(MS/MS 扫描)进行测定。在离子阱 (IT ) 质谱仪中,离子的选择、片段裂解和片段分析都是在同一池子(离子阱)进行的。实际上,MS/MS 仪器可以进行 MS 和 MS/MS 之间的切换。当离子选择和碎片裂解关闭时,MS 就可以进行扫描。MS 扫描可以检测所有的肽段离子和进行片段裂解。
电喷雾对无机盐非常敏感,因此肽段样品上样之前必须进行脱盐处理。最有效的一种方法是使用反相高效液相色谱法(RP- HPLC) 。反相高效液相色谱法不仅可以去除盐类 ,而且还可以纯化和浓缩肽段样品。这就是为什么液相色谱- MS/MS (LC-MS/MS)是蛋白质组学中最常见的配制。由于电喷雾技术跟样品浓度高度相关,色谱柱的直径是主要参数,减少柱子的直径可以提高灵敏度 [7] 。目前用直径 50~100 μm 的纳米级色谱柱来提高质谱仪进样口的肽段浓度。此外,利用纳米级色谱柱将流量减少到 150~300 nl/min 也可以提髙电喷雾的有效性。
在过去的十几年中,nanoLOMS/MS 技术已被广泛使用。在大多数质谱实验室,它已成为一种普遍的技术。由于电喷雾电离(ESI) 不同于 MALDI 电离过程,因此分析出的肽段结果有所不同。这些差异可以解释为什么 nanoLC- MS/MS 是 MALDI-TOF 质谱分析的一个有效补充 [ 9,10 ]。
2. 材料
2.1 胰蛋白酶消化
( 1 ) 天然聚丙烯聚合酶链反应(PCR ) 的 12 联管(0.2 ml/管)和相应的冷冻贮存盒架子(VWR International , Fontenay - sous - Bois, France)。
( 2 ) 多通道移液枪(YWR International ) 。
( 3 ) Eppendorf Repeater 分液器和 Combitips 分液管(VWR International ) 。
( 4 ) Eppendorf 热混合器 Thermom ixer ( Eppendorf, LePecq,France ) 和用于加热联管的模块加热块(VWR International ) 。
( 5 ) 修饰胰蛋白酶(Promega,Madison,USA ) 母液(200 ng/μl ) 。
( 6 ) 脱色液:10% 乙酸的 40% 甲醇(用 dH2O 配制)。
( 7 ) 清洗液:50 mmol/L NH4HCO3 ( 用 dH2O 配制,母液 1 mol/L ) 。
( 8 ) 消化缓冲液:25 mmol/L NH4HCO3,20% 甲醇(用 dH2O 配制)。
( 9 ) 收缩液:100% 乙腈。
( 10 ) 萃取液:0.5% 三氟乙酸(TFA ),40% 乙腈,10% 甲醇(用 dH2O 配制)。
2.2 高效液相色谱仪(NanoLC)
( 1 ) NanoHPLC 终极系统:Famos-Switchos-Ultimate ( Dionex-LC Packings,France) 。
( 2 ) C18 反相柱:300 μm X 5 mm ( trap column ) 和 75 μm X 10 cm ( analyticalcolumn ; PEPMAPC 18,Dionex LC- Packings)。
( 3 ) 流体管路接头(fitting) 和管路(tubing) ( Upchurch) 。
( 4 ) 熔融石英管(Upchurch) 。
( 5 ) 样品溶液:0.1% TFA,2% 乙腈(用 dH2O 配制)。
( 6 ) 装载溶剂(清洗捕获柱子,7 μl/min,见图 20-1): 0.1% 甲酸,0. 02% TFA,2% 乙腈。
( 7 ) 高效液相色谱溶剂A : 0.1% 甲酸,2% 乙腈(用 dH2O 配制)。
( 8 ) 高效液相色谱溶剂B : 用 95% 乙腈和 5% dH2O 配制的 0.1% 甲酸。
2.3 质谱仪(离子阱)
( 1 ) 纳米级喷针 360/20/10,未加涂层(New Objective,Woburn ) 。
( 2 ) 离子讲质谱仪,LCQ XP Plus ( Thermo Electron,Courtaboeuf,France) 。
( 3 ) 数据采集软件,Xcaliburl. 3 ( Thermo Electron) 。
( 4 ) 数据处理软件,BioWorks 3.1 ( Thermo Electron) 。
4. 注释
( 1 ) 建议以一系列代表性的样品在加或不加 DTT/IAA 情况下验证还原和烷基化反应的效率(或无效性)。检查已鉴定肽段的数量,角蛋白的污染,和是否存在羧胺甲基化半胱氨酸(+ 57 Da) 。
( 2 ) SpeedVac 会导致颗粒和干燥蛋白的溶解性减小,降低肽段的回收率。30 μl 的提取物在室温下需要过夜蒸发或在 4°C 蒸发 1~2 天。
( 3 ) 液体接头应紧固耐用,足以运行数百个样品而不会有任何问题(在堵塞的情况下除外)。
( 4 ) 液体接头的电极丝必须抗氧化。可以使用电泳仪用的销金线。
( 5 ) 分流是一种有效的方法,可以减少泵、泵管、混和器的死体积。另外,流速取决于来自分流管(nanoLC 系统的一般为 10 μm 内径)和柱子的压力。这些压力的任何变化都会改变流速。利用活动分流系统可以改善这一性能。
( 6 ) 在 m/z 大范围进行全质谱扫描是 “低分辨率” 扫描。在 m/z 短范围内(10 m/z 集中在父本离子)进行变焦 MS 扫描是 “高分辨率” 扫描。这种扫描可以测定肽段的电荷和质量。这里为 MS/MS 选择了两个离子。选定离子的数量取决于肽段的洗脱时间、肽段混合物的复杂性、每次扫描的速度以及仪器的灵敏度。对于复杂混合物,不使用 MS ZoomScans,增加选定离子的数量(最多 5 个) 。这就会增加待分析肽段的数量。
( 7 ) 纳流喷雾源安装摄像头,可以让用户看到喷头和喷雾,便于调整和故障排除 。当 LC 泵开始运行接通电压后,液滴必须喷出,喷头上不能有任何液体。
( 8 ) 在 http://www. abrf. org/ABRFNews/1996/Septemberl996/sep96iontrap . html 可找到有关 IT 和 Qz 对除去低质量的影响较为全面的教程。
( 9 ) 动态排除列表中对应的是父本离子的质量,这些是在最后几秒钟检测得到的。这可以防止对相同的父本离子进行重复 MS/MS 扫描。此外,这种设置还可以分析共洗脱时流出的低丰度的离子。
( 10 ) EST 是 DNA 序列(一般含 200~500 个核苷酸),是在系统地对 cDNA 文库的两端进行测序时所产生的。EST 重叠群是 EST 簇的共有序列。单一序列是跟任何 EST 没有相似性的 EST。EST 重叠群和 EST 单一序列通常分组;相应的数据库称作假定独立基因(tentative unique genes,TUG ) 。TUG = TUC + TUS。 TUC 和 TUS 分别是假定独立重叠群( tentative unique contigs) 和假定独立单一序列(tentative uniquesinglets) 的缩写 。
( 11 ) 参见 IPI ( 国际蛋白索引)ftp : //ftp . ebi. ac. uk/pub/databases/IPI/current/。新的标准有最新数据库:ftp ://ftp.ncbi. nih.gov/blast/db/FASTA / 和植物的蛋白序列数据库: http://www.plantgdb.org/download.p hp。
( 12 ) 选择修改变量会增加索引数据库的大小,延长查询时间并降低相关性得分。除了蛋氨酸氧化外,在第一轮查询时不必修改任何变量,以降低鉴定假象。
( 13 ) 在 MS/MS 分析中,角蛋白和胰蛋白酶肽段的少量污染不算什么问题。只有当它们的峰是最强的峰时才成为难题,在这种情况下感兴趣的肽段对应的小峰,可能会被屏蔽而不能分析。
参考文献
1. Washburn, M . P., Wolters, D., and Yates, J. R. 3rd. (2001) Large- scale analysis ofthe yeast proteome by multidimensional protein identification technology. Nat.Biotechnol. 1 9 , 242-247.
2. Froehlich, J. E., Wilkerson, C. G., Ray, W . K., et al. (2003) Proteomic study of theArabidopsis thalianachloroplastic envelope m e m b r a n e utilizing alternatives totraditional two-dimensional electrophoresis. J. Proteome Res.2, 4 1 3 -42 5 .
3. Lin, D., Tabb, D. L., Yates, J. R. Ill (2003) Large-scale protein identificationusing mass spectrometry. Bioch. Biophys. Acta 16 4 6, 1- 10.
4. Ferro, M., Salvi, D., Brugiere, S., et al. (2003) Proteomics of the chloroplast envelope m e m b r a n e s from Arabidopsis thaliana. Mol. Cell. Proteomics2, 325-345.
5. Carter, C., Pan, S., Zouhar, J., Avila, E. L., Girke, T., and Raikhel, N. V. (2004)T he vegetative vacuole proteome of Arabidopsis thalianareveals predicted andunexpected proteins. Plant Cell. 16, 3285 -3303.
6. Friso, G., Giacomelli, L., Ytterberg, A. J., et al. (2004) In-depth analysis of thethylakoid m e m b r a n e proteome of Arabidopsis thalianachloroplasts: n e w proteins,n e w functions, and a plastid proteome database. Plant Cell. 16, 478-499.
7. Abian, J., Oosterkamp, A. J., and GelpI, E. (1999) Comparison of conventional,narrow-bore and capillary liquid chromatography/mass spectrometry forelectrospray ionization mass spectrometry: practical considerations. J. MassSpec from.34, 244-254.
8. Wilm, M . and Man n , M . (1996) Analytical properties of the nanoelectrospray ionsource. Anal. Chem.68, 1-8.
9. Bodnar, W . M., Blackburn, R. K., Krise, J. M., and Moseley, M . A. (2003)Exploiting the complementary nature of L C / M A L D I / M S / M S and LC/ESI/MS/M S for increased proteome coverage. J. Am. Soc. Mass Spectrom. 1 4 , 971-979.
10. Lim, H., Eng, J., Yates, J. R. 3rd, et al. (2003) Identification of 2D-gel proteins: acomparison of M A L D I / T O F peptide mass mapping to m u L C - E S I tandem massspectrometry. J. Am. Soc. Mass Spectrom. 1 4 , 957-970.
11. Schagger, H. and von Jagow, G. (1991) Blue native electrophoresis for isolationof m e m b r a n e protein complexes in enzymatically active form. Anal. Biochem.1 99 , 223-231.
12. Davis, M . T. and Lee, T. D. (1997) Variable flow liquid chromatography-tandemmass spectrometry and the comprehensive analysis of complex protein digest mixtures. J. Am. Soc. Mass Spectrom.8, 1059-1069.
13. Schlosser, A. and Volkmer-Engert, R. (2003) Volatile polydimethylcyclosiloxanes in the ambient laboratory air identified as source of extreme backgroundsignals in nanoelectrospray mass spectrometry. J. Mass. Spectrom.38, 523-525.
14. Roepstorff, P. and Fohlman, J. (1984) Proposal for a c o m m o n nomenclature forsequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601.
15. Yates, J. R. 3rd, Eng, J. K., and M c C o r m a c k , A. L. (1995) Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequencesin nucleotide databases. Anal. Chem.67, 3202-3210.
16. Eng, J., M c C o r m a c k , A. L., and Yates, J. R. III. (1994) A n approach to correlatetandem mass spectral data of peptides with amino acid sequences in a proteindatabase. J. Am. Soc. Mass Spectrom.5, 976-989.