如何利用转录组数据进行 GSEA 分析?
丁香园
今天,我们从真实数据出发,利用已发表文献中的数据进行实战。
如何找到 GSEA 所需要的数据?
我们选择的文献是安秀丽老师团队最近在 Blood 上发表的一篇题为《Identification and transcriptome analysis of erythroblastic island macrophages》的文章。感兴趣的小伙伴可以详细读一下这篇文章,而我们目前要做的就是利用其转录组数据进行 GSEA 分析。
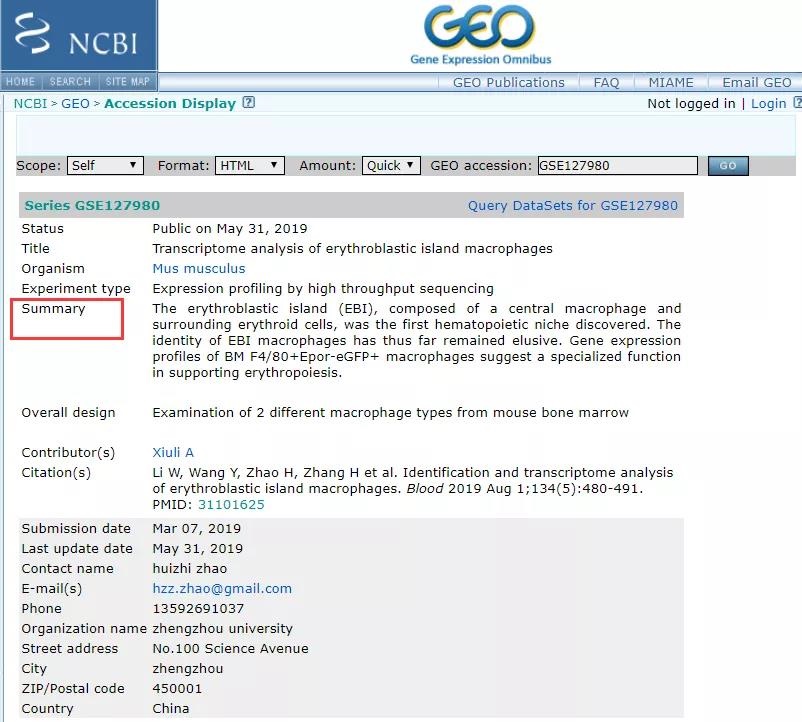
首先,我们根据其 GSE 号在 NCBI 中的 GEO 数据库中找到该文章中的转录组数据,数据的具体信息如下图所示,通过「Summary」我们可以简单了解该文章的主要内容,即鉴定并分析血岛中巨噬细胞的特征。
我们看到文章中的样本数为 6 个,三个为 GFP 阳性,三个为 GFP 阴性,这两类样本代表血岛中的巨噬细胞和非血岛中巨噬细胞。这种样本组成也类似于「Case」和「Control」,是大多数研究中所采用的样本组成。
在「Supplementary file」部分,我们看到作者为我们提供了 FPKM 数据矩阵,而这正是我们做 GSEA 需要的输入数据,所以说该数据是我们需要下载的。
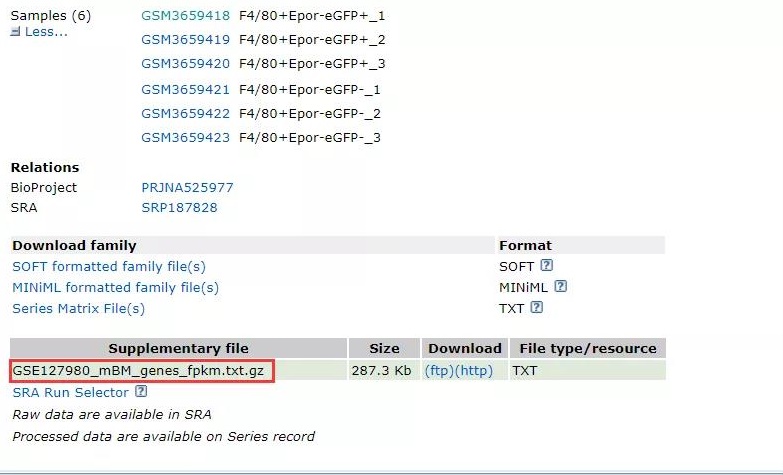
我们先来看看数据结构:每一列的表头为样本名称;每一行的表头为基因名称。
数据前三列为 GFP 阴性转录组数据,后三列为 GFP 阳性转录组数据。需要注意的是,该数据集的格式为文本文档,即 txt 结尾的格式。

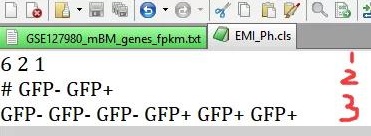
除了上述的表达矩阵,我们还需要一个解释样本类型的文件,如下图所示,文件格式为 cls 结尾,文件组成包括三类:
1)共 6 个样本,2 种类型,最后那个 1 为约定俗成的格式,姑且理解为每个样本都是独一无二的吧;
2)#开头,空格符之后是两种类型样本的描述;
3)样本的排列顺序,和前面提到的表达矩阵一致,前三个为 GFP-;后三个为 GFP+。
如何导入数据?
1、打开我们已经安装好的 GSEA 软件,点击左上角的「Load data」来导入我们的数据。
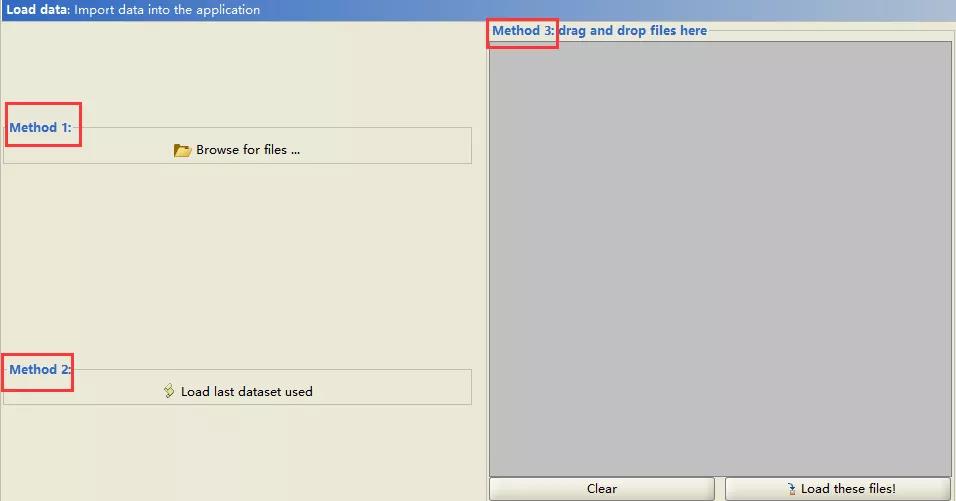
之后,我们可以有三种方式导入数据:
浏览文件夹;
直接将文件拖入;
重新导入上次用过的数据。
由于我们这次是新数据,所以选择前两种的其中一种导入即可。
选择我们下载的表达矩阵数据和我们自己创建的样本类型解释文件进行导入,此时我们可以看到以下界面:成功导入 2 个文件,无错误。
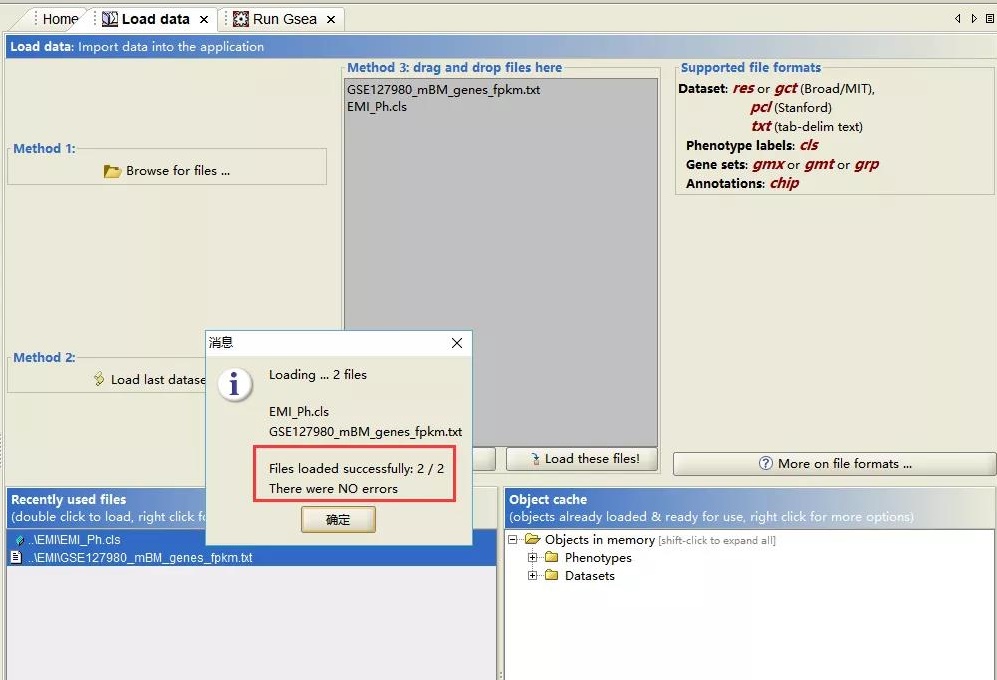
2、进行第二步「Run GSEA」。在该步骤中,会出现一个新的页面,需要我们自行设置一些参数,这也是进行 GSEA 最为关键的一步。
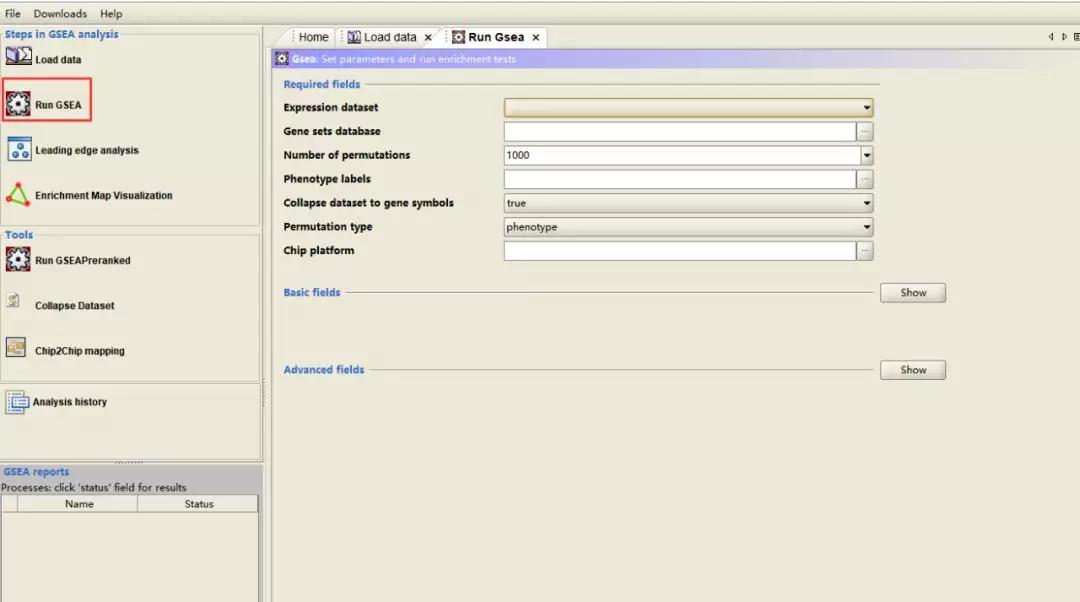
「Expression dataset」选择的是我们从数据库中下载的 FPKM 表达矩阵;
点击「Gene sets database」的最末尾处,会出现非常多的数据集可供选择,在这里,我们选择 Hallmarks 基因集,该基因集包含了常见的生物学现象及相关通路,可作为 GSEA 数据分析的初筛;
紧接着,在「Phenotype labels」中选择我们上传的样本解释文件;
剩下的两行分别选择「false」和「gene_set」。

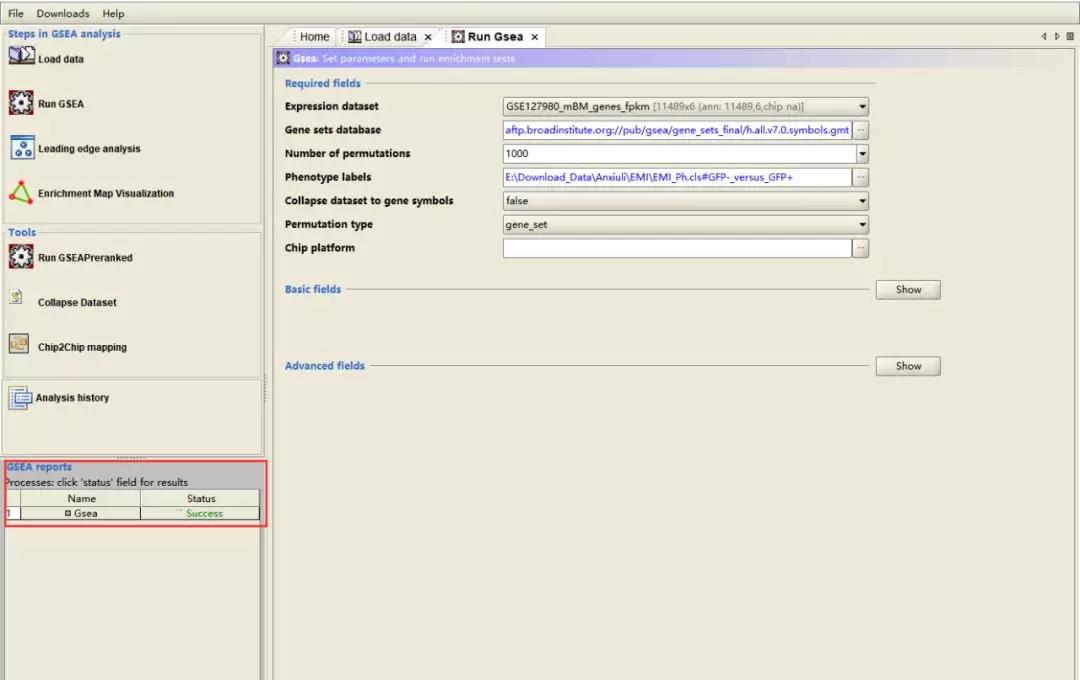
3、设置好以上参数之后,点击最下方的「Run」,此时软件界面的左下方为显示运行的状态「Running」;等到运行结束之后,状态会变成「Success」。此时,点击「Success」,即可看到运行的结果。

如何进行 GSEA 结果分析?
下图为 GSEA 运行结果的网页版,我们可以看到整体的结果概述:
多少基因集显著富集在了哪种表型;
有多少是 FDR 小于 25% 的基因集;
有多少是 P 值小于 1% 的基因集;
有多少是 P 值小于 5% 的基因集
……

详细来看,我们看到 49 个基因集中有 24 个在 GFP 阴性样本中上调,20 个 FDR 小于 0.25,15 个基因集的 P 值小于 0.05。
点击「Snapshot」可以进一步查看每个基因集的富集情况。

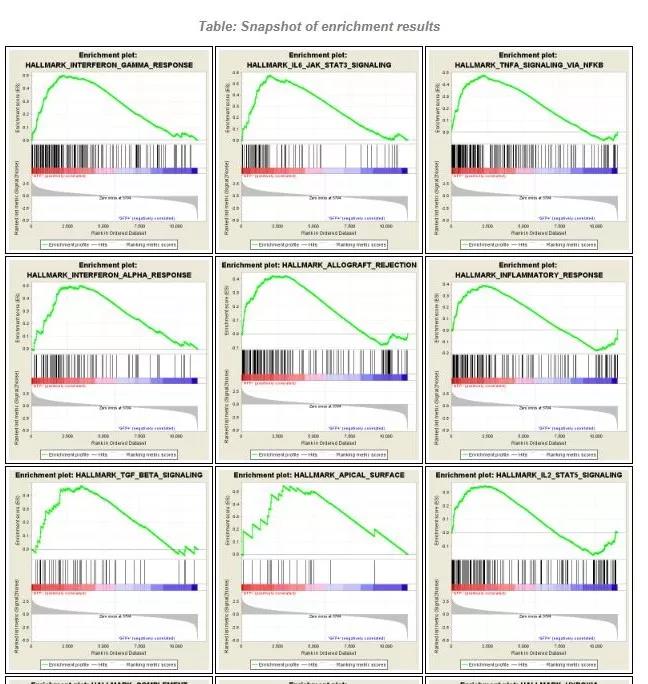
上图为整体概貌,要想了解每个基因集的具体富集程度和显著性,可以点击相应的基因集。
如下图,该图也可直接用于发文章,另外在左上方我们可以看到具体信息,其中基因集名称,NES 值,FDR 值,这三者是尤为重要的,在发文章时需要在 GSEA 富集图上标出。
NES 表示富集的程度,FDR 为其是否具有统计学差异,即显著性。
具体到本次数据分析中,我们可以解读为 Interferon-Gamma-response 信号通路在 GFP-样本中显著富集,即相对于血岛中的巨噬细胞,非血岛巨噬细胞的 Interferon-Gamma-response 通路更强。
如果小伙伴对其他通路也感兴趣,也可以一一点击查看哦。

到这里,我们就对一套转录组数据进行了完整的常规 GSEA 分析,希望该教程可以帮助到你!