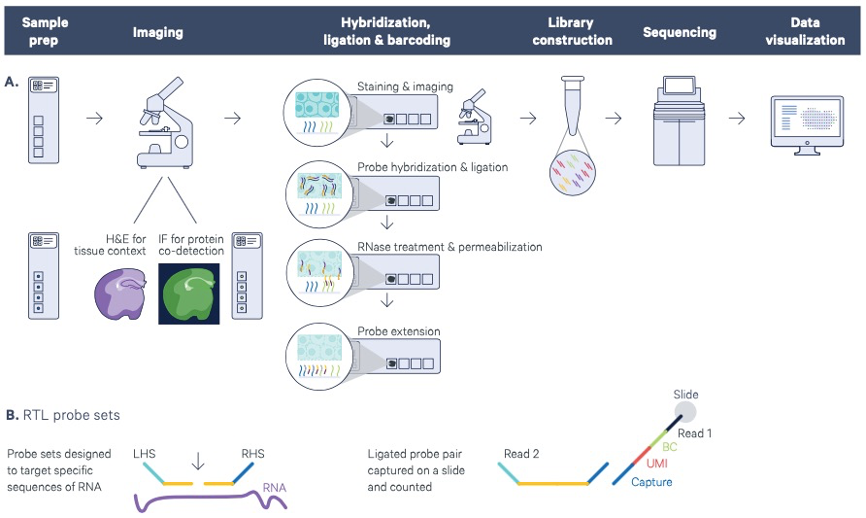
如何解析空间转录组 spot 细胞组成
上海烈冰生物医药科技有限公司
3077
科研的魅力之处,在于无穷尽的探寻生命的本底,通过单细胞测序,我们对组织器官的分子基础的研究也从初级到深层,并在单细胞层面逐渐达成一致,而空间异质性是器官功能的关键特征,细胞的位置信息更能反应细胞命运调控机制和细胞谱系发生过程。因此时间和空间即像组织器官的AB面,而不可否认的是,此时空间坐标的记录,像一个还未长大的娃娃,虽未触及分子基础研究的深层,但仍在努力攀登探寻生命本底的下一个高峰,我们结合烈冰现有实战经验和已发表的高分文献,为您介绍3种解析空间转录组spot细胞组成的分析工具。
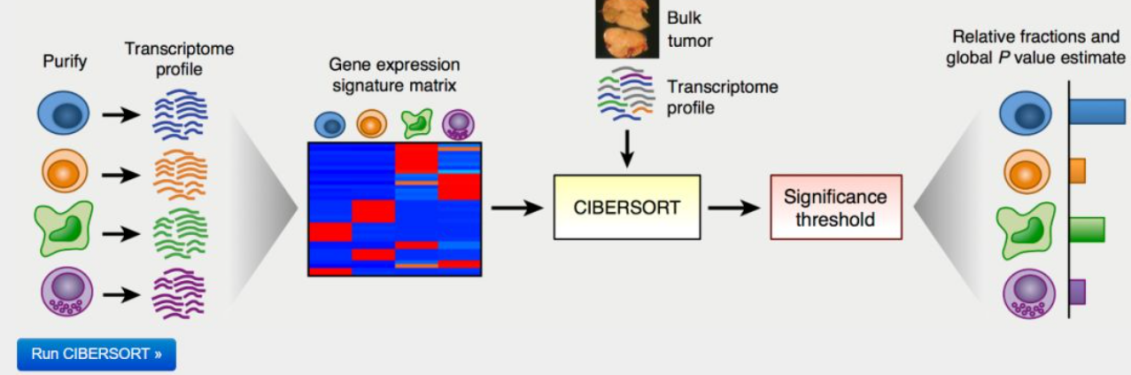
反卷积
首先基于单细胞测序数据作为背景,获得不同细胞类型的特征基因(markergene),由此,结合空转bulk数据矩阵和单细胞基因表达数据以及单细胞类型,采用Cibersort算法,该方法在2015年首次发表于《Nature Method》,是基于线性支持向量回归的原理对细胞亚型的表达矩阵进行去卷积的一个工具,能够反向推算空间转录组每个spot区域中的基因表达情况,估计混合细胞群体中存在的细胞类型和细胞占比,并在切片上进行展示(以百分比形式展示)。
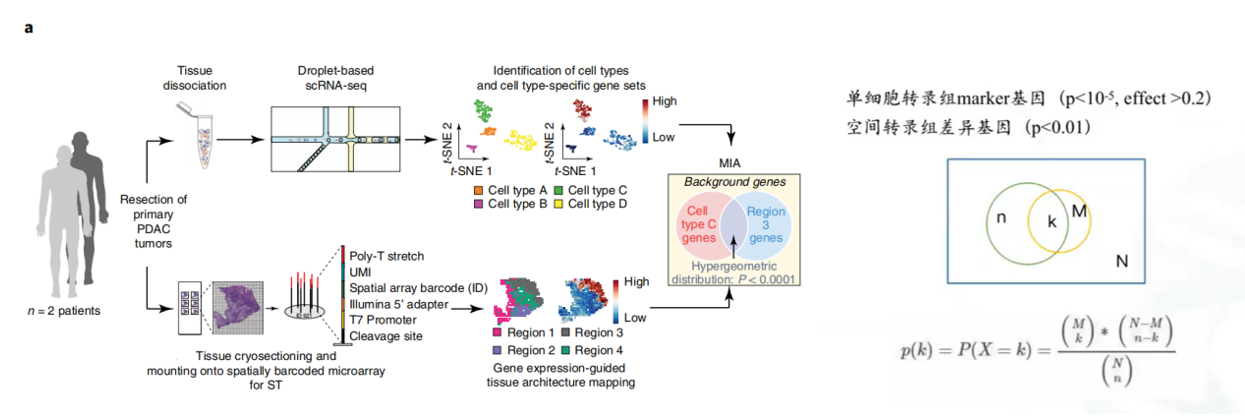
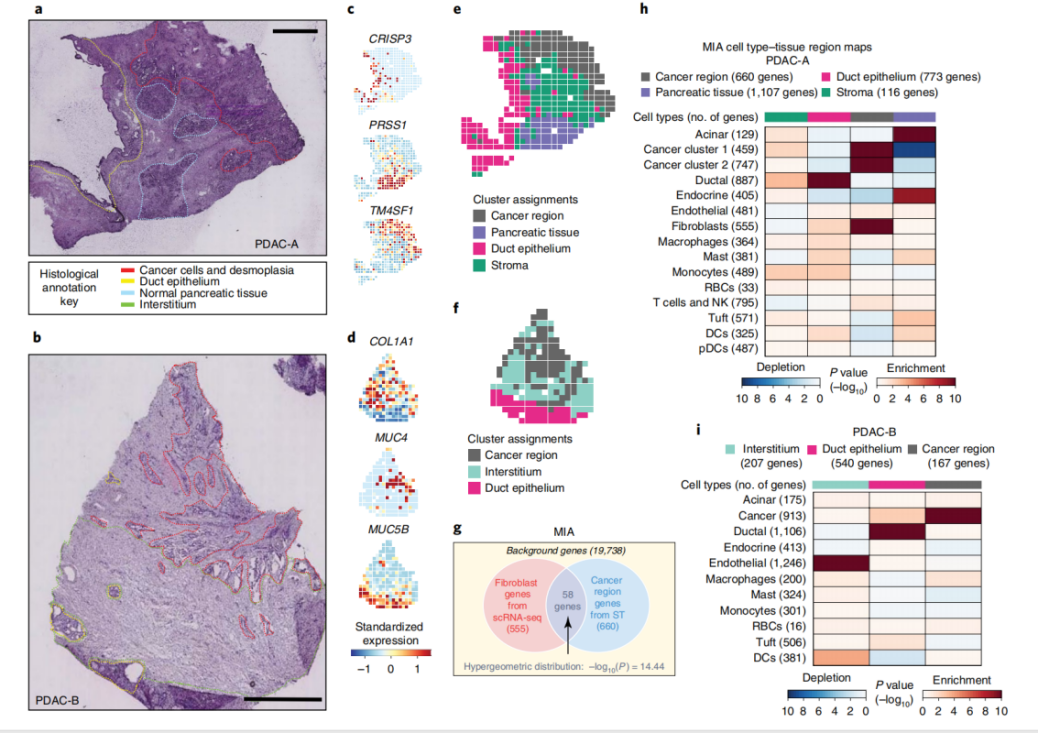
多模式相交(Multimodal Intersection Analysis,MIA)
该方法基于单细胞转录组基因表达数据和空转的基因表达矩阵,采用超几何分布检验,分别得到单细胞测序和空间转录组测序的两个genelist,对于单细胞和空转的某个区域中的markergene信息分析,推算空间转录组区域内的细胞组成。该方法见于2020年发表于《Nature Biotechnology》的一篇整合空间转录组和单细胞转录组分析的文章。
单样本基因集富集分析(ssGSEA)
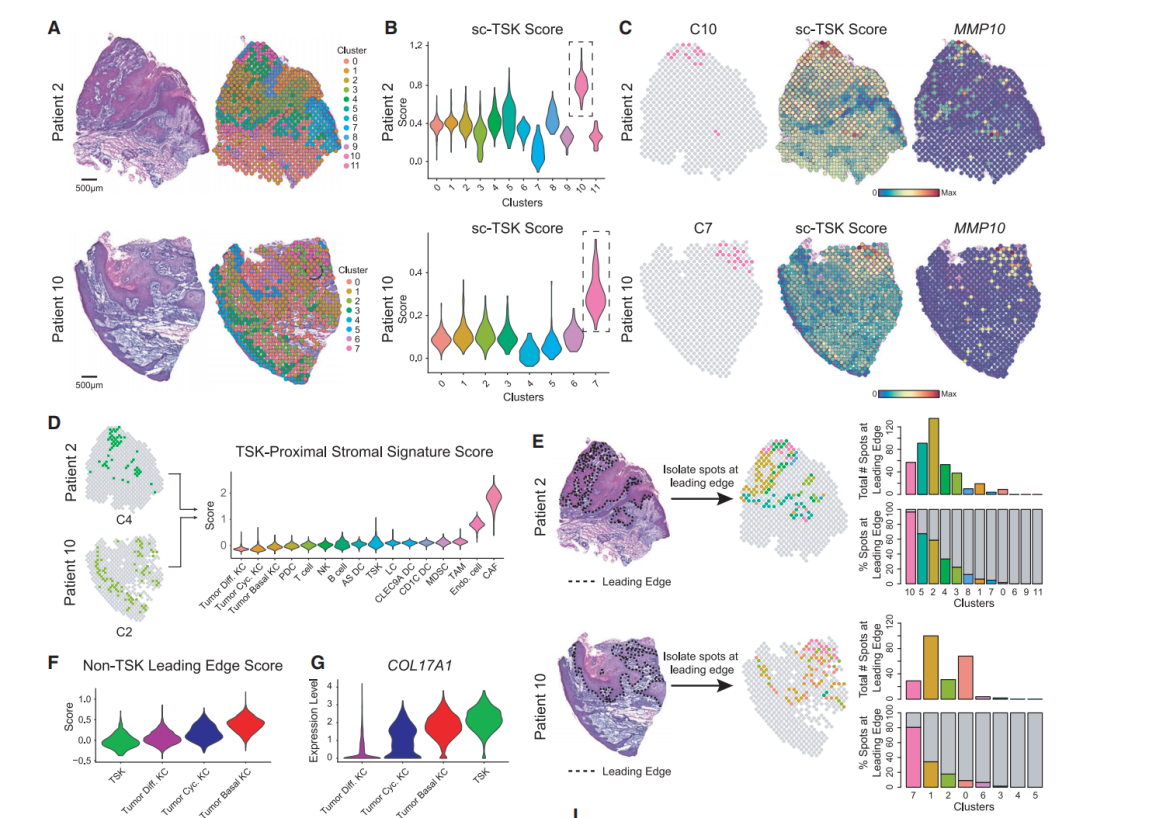
基因集富集分析(GSEA)相信大家都比较熟了,ssGSEA即single cell GSEA,这里的单样本在单细胞中看作单个细胞,而空转中可看作单个spot,像bulkRNA测序一样,空转每个spot可看作一组小型的bulkRNA数据。
单独用ssGSEA,其原理是基于不同的geneset或pathway在空间转录组中的表达情况,对比任意划分空间区域内基因集激活情况,而在细胞组成推断中,我们也可以基于已有的单细胞数据,获得细胞的特征基因集(markergene list),进一步集合空转表达矩阵,通过score打分,推算获得细胞再空间上可能的定位信息,做细胞定位工作。该方法可参考2020年7月23日,发表在Cell上的一篇人鳞状细胞癌的空间转录组文章。
反卷积
首先基于单细胞测序数据作为背景,获得不同细胞类型的特征基因(markergene),由此,结合空转bulk数据矩阵和单细胞基因表达数据以及单细胞类型,采用Cibersort算法,该方法在2015年首次发表于《Nature Method》,是基于线性支持向量回归的原理对细胞亚型的表达矩阵进行去卷积的一个工具,能够反向推算空间转录组每个spot区域中的基因表达情况,估计混合细胞群体中存在的细胞类型和细胞占比,并在切片上进行展示(以百分比形式展示)。
多模式相交(Multimodal Intersection Analysis,MIA)
该方法基于单细胞转录组基因表达数据和空转的基因表达矩阵,采用超几何分布检验,分别得到单细胞测序和空间转录组测序的两个genelist,对于单细胞和空转的某个区域中的markergene信息分析,推算空间转录组区域内的细胞组成。该方法见于2020年发表于《Nature Biotechnology》的一篇整合空间转录组和单细胞转录组分析的文章。
单样本基因集富集分析(ssGSEA)
基因集富集分析(GSEA)相信大家都比较熟了,ssGSEA即single cell GSEA,这里的单样本在单细胞中看作单个细胞,而空转中可看作单个spot,像bulkRNA测序一样,空转每个spot可看作一组小型的bulkRNA数据。
单独用ssGSEA,其原理是基于不同的geneset或pathway在空间转录组中的表达情况,对比任意划分空间区域内基因集激活情况,而在细胞组成推断中,我们也可以基于已有的单细胞数据,获得细胞的特征基因集(markergene list),进一步集合空转表达矩阵,通过score打分,推算获得细胞再空间上可能的定位信息,做细胞定位工作。该方法可参考2020年7月23日,发表在Cell上的一篇人鳞状细胞癌的空间转录组文章。