简介
通过将预先组装好的 Cas9 核糖核蛋白 (Cas RNP)传递到细胞内进行基因组编辑正成为一种日渐流行的方法,适用于难以通过常规质粒和病毒转载法进行基因操作的细胞类型。Cas9 RNP 编辑方法高效、精确、可重复,并且不含遗传物质。但它仅能在细胞中存在很短时间,因此限制了其编辑活性。本实验方案描述了通过在大肠杆菌 (Escherichia coli)中异源表达和提纯制备重组化脓性链球菌 (Streptococcus pyogenes) Cas9 (SpCas9)的方法,以及通过体外转录和 PAGE 纯化合成 CRISPR 引导 RNA 的方法。
SpCas9 是首个被发现的 CRISPR Cas9 (Jinek et al.2012),也是基因组编辑应用中最具特征性的 Cas 酶之一。
通过用本文中的 Cas9 RNP 合成法,我们阐述了一种通过电穿孔在原代人类T细胞和自然杀伤 (NK)细胞中进行高效基因组编辑的方法,以及通过聚乙二醇介导的转化方式在真菌和植物中进行基因组编辑的方法。我们的 Cas9 RNP 制备实验方案具有一致性,并且可以直接用于其他细胞类型和生物体的基因组编辑。
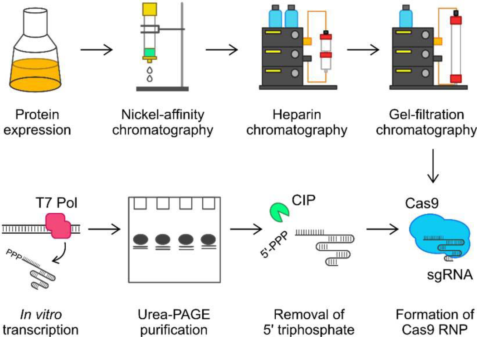
图 1 的 Cas9 核糖核蛋白传递到细胞内基因组编辑流程图
原理
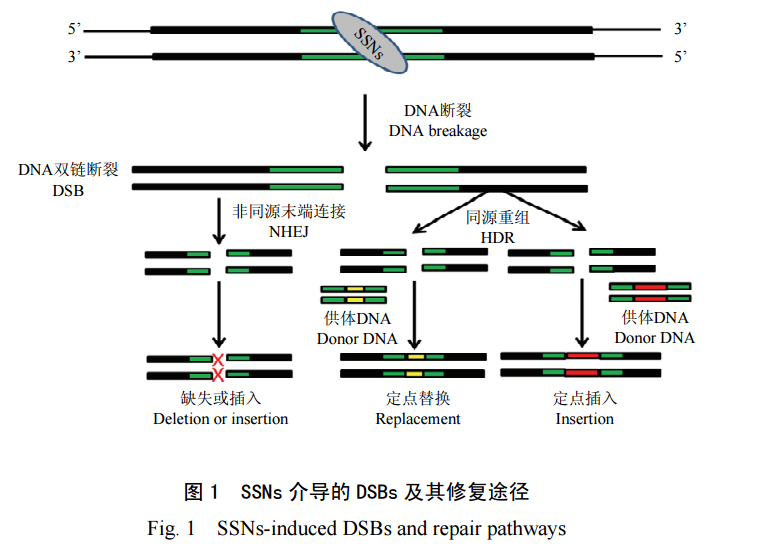
CRISP-Cas9 基因组编辑是一种广泛应用于研究生物技术、农业和医学领域的革命性技术。Cas9 核酸酶作为「分子剪刀」,可在引导 RNA 的间隔序列所指定的部位诱导目标 DNA 双链断裂,从而创造了一个通过 DNA 修复途径修改基因组序列的独特机会。
Cas 9 引导 RNA 的原生形式由两个 RNA 分子组成:一个 CRISPR RNA (crRNA)和一个反式激活crRNA (tracrRNA) (Jinek et al.2012)。研究者可用 crRNA 和 tracrRNA 设计生成一个单一的引导 RNA (sgRNA)融合分子,便于体外和体内的应用 (Jinek et al.2012)。通过传递预先组装好的 Cas9 RNP 进行基因组编辑是一种既稳健又精确的方法,当需要非常短时的编辑活性时,这种方法比标准的质粒和病毒方法更有优势。
我们可以提前合成和检验 Cas9 蛋白和引导 RNA,并将其稳定地储存在 -80℃ 冰箱中,以便在需要时用于进行基因组编辑。在科研和临床应用中,这些特性使其成为生产符合良好生产规范标准的基因组工程免疫细胞的理想选择。
在本文中,我们对重组 SpCas9 的异源表达、纯化以及 sgRNA 的酶促合成的操作程序进行了描述。由于针对的是在原代免疫细胞中的应用,这些细胞对内毒素和外源性 RNA 更为敏感,因此我们的实验方案更加侧重于 Cas9 RNP 的高纯度和低毒性。
我们的实验方案对 Savic et al. (2019)和 Nikolay et al. (2017)的实验方法进行了一些改进,包括延长了 HIS 标签以增加与镍亲和柱的结合能力,使用了温和洗涤剂以减少内毒素污染,改进了离子交换和凝胶过滤层析法,以及细胞培养中使用了经灭菌处理的蛋白质。
我们的酶法合成 sgRNA 的实验方法包括通过变性 PAGE 纯化提取全长的 sgRNA 并去除截短的 sgRNA 产物。我们还通过磷酸酶处理法去除了对人类免疫细胞具有高毒性的免疫源性 5’ 三磷酸盐基团 (Wienert et al.2018; Kim et al.2018; Huang et al.2021)。
本研究表明,我们的引导 RNA 与市售的化学合成引导 RNA 具有同样高的质量 (Huang et al.2021),但制备成本相比后者则大大降低。Cas9 和 sgRNA 制备的大致流程见图 1。
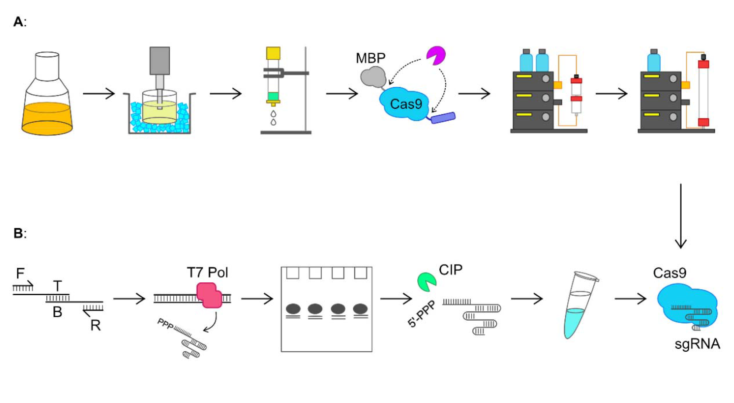
图 2. Cas9 和 sgRNA 合成和纯化方法的简略图。 (A)在大肠杆菌中表达 Cas9,并通过镍亲和层析、离子交换 (肝素柱)层析和凝胶过滤层析从大肠杆菌裂解液中提纯 Cas9。 (B) sgRNA 合成的起始步骤为以四种 DNA 寡核苷酸为原料通过重叠 PCR 组装 sgRNA DNA 模板:顶部 (T),底部 (B),正向 (F)和反向 (R)。然后用 T7 RNA 聚合酶 (T7 Pol)在体外转录 DNA 模板,生成 sgRNA。通过尿素-PAGE 电泳对 sgRNA 进行纯化后,用小牛肠道磷酸酶 (CIP)处理去除 sgRNA 的免疫源性 5’ 三磷酸基。将得到的 Cas9 和 sgRNA 混合形成 Cas9 核糖核蛋白 (Cas9 RNP),以用于基因组编辑。
材料与仪器
步骤
A、SpCas9 蛋白的纯化和表达
1、SpCas9 在大肠杆菌 Tuner (DE3) 细胞中的过量表达
a、制备大肠杆菌培养基和储备原液
1)准备50 mL 的 Terrific Broth 培养基。在室温下保存。
2)在一个 250 mL 的锥形瓶中加入 100 mL 的 LB+ 甘油培养基用于初始培养。在室温下储存,并在一周内使用。
3)在三个 5 L 的 Ultra-Yield 培养瓶分别加入 1000 mL 的 LB+ 甘油培养基,用于大量培养。在室温下储存,并在一周内使用。
4)制备 20× 改良 M9 缓冲液。在室温下最长可储存一年。
5)制备 1 M IPTG 原液和 50 mg/mL 卡那霉素溶液。储存在 -20℃ 下,最长可保存两年。
6)制备含有 50 μg/mL 卡那霉素的LB琼脂培养板。储存在 4℃ 下,最长可保存一个月。
7)制备裂解缓冲液。储存在 4℃ 下,最长可保存一个月。
b、大肠杆菌的转化 (第 1 天)
1)在冰上解冻化学感受态 Tuner (DE3)细胞。
2)在 50 μL 的感受态细胞中加入 1 ng 的 spCas9 表达质粒 (Addgene)。
3)通过轻弹混匀,并将试管在冰水孵育 20 分钟。
4)将试管在 42℃ 水浴中放置 60 秒,对大肠杆菌进行热休克处理。
5)将试管置于冰水孵育 2 分钟。
6)向大肠杆菌中加入 500 μL 的 Terrific Broth 培养基,轻弹混匀。
7)将大肠杆菌在 37℃ 下摇晃孵育 1 小时,以恢复活性。
8)将 100 μL 的细胞液种到含有 50 μg/mL 卡那霉素和无菌玻璃微珠的 10 cm LB 琼脂培养板上,并待培养板完全干燥。
9)倒出玻璃微珠,将培养板在 37℃ 下孵育 18-20 小时,以使大肠杆菌形成菌落。
10)将培养板储存在 4℃ 下并在两周内使用。
注:Tuner (DE3)菌株无法长期稳定的维持 Cas9 质粒。我们建议表达每一批 Cas9 时都要用新鲜转化的 Tuner (DE3)菌株。
c、设置大肠杆菌的起始培养 (第 2 天)
1)在 100 mL LB+ 甘油培养基中加入 2.5 mL 的 20× 改良 M9 缓冲液和 102 μL 的 50 mg/mL 卡那霉素溶液,重新组成完全培养基。
2)用无菌移液器吸头从 LB 琼脂培养板上挑出 4-5 个单菌落,接种到完全培养基中。
3)将起始培养物以 200 rpm 摇速在 30℃ 下摇动培养 20 小时。注意不要在更高温度下长时间培养,避免大肠杆菌的过度生长。
d、spCas9 在大规模培养基中的表达 (第 3 天)
1)将 100 mL 的起始培养液分装在两个 50 mL 的锥形管中。
2)以 4000× g 转速在 25℃ 下离心 10 分钟,收集大肠杆菌,并弃去培养基。
3)通过涡旋将两管大肠杆菌颗粒重新悬浮在总量 15 mL 的 Terrific Broth 培养基中。
4)在 1000 mL LB+ 甘油培养基中加入 25 mL 的 20× 改良 M9 缓冲液和 1030 μL 的 50 mg/mL 卡那霉素溶液,重新组成完全培养基。
5)在每个装有完全培养基的培养瓶中加入 5 mL 大肠杆菌悬液。共设置三个这样的培养瓶。
6)将培养瓶以 200 rpm 摇速在 37℃ 下摇动培养。
7)通过从中吸取 500 μL 培养物并在 600 nm 处测定吸光度来监测大肠杆菌密度。
8)当 600 nm 处的吸光度达到 1.5 时,将培养瓶从培养箱中取出,在冰水中孵育 10 分钟以降低温度。中间每隔 2~3 分钟手动摇瓶。
9)然后在其中加入 IPTG 致使其终浓度为 500 μM。
10)将培养瓶在 16℃ 下以 200 rpm 的摇速摇动孵育 18 小时,以诱导 Cas9 的表达。
e、大肠杆菌的收获和储存 (第 4 天)
1)将大肠杆菌培养物从培养瓶中转移到 1 L 聚丙烯离心瓶中。
2)离心前在电子秤上平衡各离心瓶,通过加去离子水调整重量。
3)以 3500× g 转速在 4℃ 下离心 15 分钟,收集大肠杆菌,并弃去培养基。
4)在每个培养瓶中加入 70 mL 冰冷的裂解缓冲液重悬大肠杆菌颗粒。
5)通过涡旋+移液器吹打的方式打散大肠杆菌颗粒。将重悬后的大肠杆菌置于冰上。
6)将大肠杆菌悬液合并在一起并立即进行步骤2b,即通过超声处理裂解大肠杆菌。
7)或者,也可以将大肠杆菌悬液等分到 50 mL 的锥底管中,在 -80℃ 下冷冻保存。注意每个锥底管中装量不要超过 45 mL,以免在冷冻过程中锥底管发生破裂。
8)然后将锥底管转移到 -80℃ 冷冻细胞。冷冻过程会部分破坏大肠杆菌细胞膜,有助于增加细胞裂解度。冷冻的大肠杆菌悬液在 -80℃ 下至少可以稳定保存一年。
2、SpCas9 的纯化
a、制备纯化缓冲液和储存原液
1)制备用于镍亲和层析的裂解缓冲液、洗涤缓冲液和洗脱缓冲液。储存在 4℃ 下,最长可保存一个月。
2)制备用于肝素离子交换层析的 IEX A 缓冲液和 IEX B 缓冲液。储存在 4℃ 下,最长可保存一个月。
3)制备凝胶过滤缓冲液。储存在 4℃ 下,最长可保存一个月。
4)制备 100 mM PMSF 储存原液。储存在 -20℃ 下,最长可保存一年。
5)制备 10 mg/mL DNase I (来自牛胰腺)溶液。将其按 1 mL 等份分装,在液氮中速冻,并储存在 -80℃ 下,最长可保存 2 年。
6)制备 5 N NaOH+0.1 mM EDTA 溶液。在 4℃ 下保存,最长可保存一年。
b、通过超声处理裂解大肠杆菌 (第 5 天)
1)在冰冷自来水中解冻 6 管冷冻大肠杆菌悬液 (约 300 mL)。
2)每隔 5 分钟将大肠杆菌悬浮液轻轻倒置一次,直到其中的冰块完全融化。
3)在解冻细胞颗粒的同时,设置一个 400 mL 的玻璃烧杯,在里面放入一个搅拌子,置于搅拌板上,并将它们置于超声探头下。
4)在室温下,将 30 mg 溶菌酶溶解在装有 30 mL 溶菌缓冲液的烧杯中。
5)将解冻后的大肠杆菌悬液倒入烧杯中。
注:解冻后的大肠杆菌悬液应该是粘稠的,因为冷冻会导致部分细胞裂解。
6)将烧杯放在冰桶的中心,用冰块包围烧杯,以使超声处理时烧杯始终处于低温中。
7)将装有烧杯的冰桶放在搅拌板上,将转速设置为 100 rpm,使大肠杆菌悬液均质化。
8)保持搅拌的同时,慢慢地将 3 mL 的 100 mM PMSF 溶液逐滴加入大肠杆菌悬液中。
9)保持搅拌的同时,向大肠杆菌悬液中加入 1 mL 10 mg/mL DNase I (来自牛胰腺)。
10)将超声探头浸入大肠杆菌悬液中,深度约为5英寸。
注:在超声处理前,大肠杆菌悬浮液应呈不透明状,并带浅黄色。
11)通过超声处理 (输出功率 40,超声脉冲每开启 20 秒后关闭 20 秒,总时长为 10 分钟)裂解大肠杆菌,同时不断搅拌,使细胞裂解液均质化,并带走超声探针产生的热量。
12)将约 37 mL 的大肠杆菌裂解液转移到离心管中。根据需要使用多个离心管。不要装得过满,以免在离心过程中发生泄漏。
13)离心前在电子秤上平衡离心管,并通过添加裂解缓冲液调整重量。
14)在 4℃ 下以 20000× g 转速离心 30 分钟。将上清液合并到一个干净的 500 mL 玻璃瓶中,并置于冰水预冷。
15)用 0.45 μm 孔径的过滤器过滤上清液以完全去除细胞碎片。将大肠杆菌裂解液的可溶性部分收集在一个干净的 500 mL 试瓶中,并置于冰上预冷。
16)取 5 μL 可溶性裂解液,与 40 μL 的 1× NuPAGE 上样染色剂混合,将混合液在 95℃ 下加热 5 分钟,然后进行 SDS-PAGE 电泳分析。
c、镍亲和层析 (第 6 天)
1)在 50 mL 锥底离心管中用 20 mL 去离子水清洗 20 mL Ni-NTA 填料。轻轻颠倒试管进行混合。
2)在 4℃ 下以 20000× g 转速离心 5 分钟,用移液器吸去上清液。
3)将 Ni-NTA 填料重悬于 40 mL 的裂解缓冲液中。轻轻颠倒试管进行混合。
4)在 4℃ 下以 20000× g 转速离心 5 分钟,用移液器吸去上清液。
5)用移液器将 Ni-NTA 填料重悬于 20 mL 的裂解缓冲液中。
6)将 Ni-NTA 浆液转移到装有可溶性大肠杆菌裂解液的 500 mL 试瓶中。
7)在瓶中放入一个搅拌子,并将试瓶放在冰桶内的搅拌板上孵育。
8)在冰上将 Ni-NTA 和大肠杆菌裂解液混合,以 100 rpm 的转速搅拌 20 分钟,以捕获 HIS 标签标记的 Cas9 蛋白。
9)将层析柱安装在一个固定支架上。在 4℃ 下进行以下步骤,让裂解液和缓冲液在重力作用下流过层析柱。
10)用移液器将 Ni-NTA 填料和裂解液浆液加载到层析柱中。弃去流穿液。
11)用 400 mL 的裂解缓冲液清洗层析柱。
12)用 400 mL 的洗涤缓冲液洗柱。
13)用移液器将洗脱缓冲液加入层析柱,按 5 mL 的固定体积将一系列馏分收集在 15 mL 的试管中。
14)通过 Bradford 法检测每份馏分,方法为从每份馏分中取 5 μL 洗脱液,在 96 孔板中与 50 μL 的 1× Bradford 试剂混合。
15)将馏分与大量的蛋白质在超滤管 (15 mL,截留分子量 100 kDa)中混合。
16)在 4℃ 下以 4000× g 转速离心 15 分钟。在管中重新加入更多的蛋白质洗脱物,并将试管轻轻倒置 4-5 次以混合内容物。
17)重复此离心过程,将洗脱液浓缩到 2 mL 的最终体积。
18)用移液管轻轻地混合浓缩后的洗脱液。
19)取 5 μL 浓缩洗脱液,与 40 μL 的 1× NuPAGE 上样染色剂一起加入到一个 1.5 mL 微离心管中并混合,将混合物在 95℃ 下加热 5 分钟后进行 SDS-PAGE 电泳分析。
20)将洗脱液转移到一个新的 50 mL 的锥底管中。
21)将 TEV 蛋白酶与浓缩后的洗脱液混合,以裂解重组 Cas9 蛋白中的麦芽糖结合蛋白融合和 12× HIS 标签。推荐使用 1:100 (w/w) 的混合比例,或通过在 280 nm 处的测量吸光度,混合 10000 单位 (1 mg)TEV 蛋白酶和 100 mg 目标蛋白。
22)将反应体系在 4℃ 下孵育过夜 (约 18 小时)。
d、肝素离子交换层析 (第 7 天)
1)取 5 μL TEV 反应混合液,与 40 μL 的 1× NuPAGE 上样染色剂一起加入到一个 1.5 mL 离心管中并混合,将混合物在 95℃ 下加热 5 分钟后进行 SDS-PAGE 电泳分析。
2)用 10 mL 的血清用移液管向 TEV 反应混合液中加入 10 mL 的 IEX A 缓冲液。方法为将移液管浸没在混合液中,轻轻旋转溶液,并同时慢慢释放 IEX A 缓冲液。
3)然后用同样的方法慢慢加入另外 20 mL 的 IEX A 缓冲液。
4)将打包混合物通过 45 μm 的注射过滤器以去除蛋白沉淀。
5)将两根 5 mL 的 HiTrap Heparin HP 肝素层析柱串联在一起。
6)用蠕动泵在 4℃ 下以约 3 mL/分钟的流速用 50 mL 的 IEX A 缓冲液平衡层析柱。
7)用蠕动泵在 4℃ 下以约 3 mL/分钟的流速将蛋白质混合液加载到层析柱上。收集流穿的组分。
8)取 5 μL 流穿组分,与 40 μL 的 1× NuPAGE 上样染色剂一起加入到一个 1.5 mL 离心管中并混合,将混合物在 95℃ 下加热 5 分钟后进行 SDS-PAGE 电泳分析。
9)用 IEX A 缓冲液 和 IEX B 缓冲液清洗 ÄKTA 层析系统,然后用 IEX A 缓冲液平衡流路。
10)将肝素层析柱连接到 ÄKTA 层析系统上,用 100 mL 的 IEX A 缓冲液以 3 mL/分钟的速度洗柱。
11)用 0%-80% 线性梯度浓度的 IEX B 缓冲液以 3 mL/分钟的流速洗脱蛋白质,洗脱液量为 150 mL。
12)如图 2 所示,合并峰值馏分。
13)将蛋白洗脱液转移到超滤管中 (15 mL,截留分子量 100 kDa)。
14)在 4℃ 下以 4000× g 转速离心 15 分钟。在管中重新加入更多的蛋白质洗脱物,并将试管轻轻倒置 4-5 次以混合内容物。
15)重复此离心过程,将洗脱液浓缩到约 5 mL 的最终体积。将浓缩后的洗脱液存放在冰水。
16)取 5 μL 浓缩的肝素层析洗脱液,与 40 μL 的 1× NuPAGE 上样染色剂一起加入到一个 1.5 mL 离心管中并混合,将混合物在 95℃ 下加热5分钟后进行 SDS-PAGE 电泳分析。
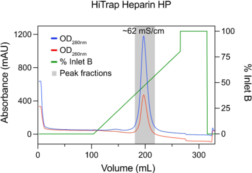
图 3. 肝素层析的洗脱曲线。
e、Superdex 200 凝胶过滤层析 (第 7 天和第 8 天)
1)在第 7 天肝素层析完成后开始,将一个 2 mL 的上样环连接到 AKTA 层析系统上,用去离子水清洗系统。
2)将 HiLoad Superdex 200 16/600 层析柱连接到 AKTA 层析系统上。在整个纯化过程中,设置压力报警和流速 (0.8-1 mL/分钟)。
3)用 250 mL 的 0.5 N NaOH+0.1 mM EDTA 溶液清洗流路。
4)用 250 mL 去离子水冲洗流路。
5)用 250 mL 的凝胶过滤缓冲液平衡流路。
6)在第 8 天,将浓缩后的蛋白质转移到注射器中。注意避免产生气泡。
7)将浓缩后的蛋白质载入 AKTA 层析纯化系统的 2 mL 上样环中。
8)以 1 mL/min 的流速将蛋白质进样到高效液相层析仪中。
9)如图 3 所示,进样后在~62 mL 的保留体积处合并峰值馏分。
10)将合并的 Cas9 馏分转移到一个超滤管中 (15 mL,截留分子量 100 kDa)。
11)在 4℃ 下以 4000× g 转速离心 15 分钟。在管中重新加入更多的蛋白质洗脱物,并将试管轻轻倒置 4-5 次以混合内容物。
12)重复此离心过程,将 Cas9 蛋白浓缩到约 2 mL。
13)取 5 μL 浓缩的 Cas9 蛋白,与 40 μL 的 1× NuPAGE 上样染色剂一起加入到一个 1.5 mL 离心管中并混合,将混合物在 95℃ 下加热 5 分钟后进行 SDS-PAGE 电泳分析。
14)用 NanoDrop Lite 分光光度计在 280 nm 处测量 Cas9 蛋白的吸光度。
15)通过 Beer 定律估计 Cas9 的摩尔浓度。重组 Cas9 的理论消光系数为 126410 M-1cm-1。
16)继续浓缩 Cas9 至 40 μM。如果浓缩过度,可用凝胶过滤缓冲液稀释 Cas9。
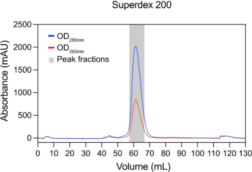
图 4. Superdex 200 层析的洗脱曲线。
f、对 Cas9 进行灭菌、冷冻和分析 (第 8 天)
1)在用 400 μL 的凝胶过滤缓冲液预湿润离心过滤器 (0.2 μm)。
2)在 4℃ 下以 12000× g 转速离心 1 分钟。弃去流穿液。
3)将 Cas9 蛋白转移到离心过滤装置上,每管 400 μL。
4)在 4℃ 下以 12000× g 转速离心 1 分钟。弃去过滤管。
5)将 Cas9 等份分装在 200 μL 的 PCR 管中 (每管 11 μL)。每次移液后立即将装好的 PCR 管在液氮中速冻。
6)将分装好的 Cas9 样本保存在 -80℃ 冰箱中。Cas9 蛋白至少可稳定保存一年。应避免反复冷冻和解冻。
7)用双蒸水将 10× Tris-甘氨酸-SDS 电泳缓冲液稀释至 1×。
8)进行电泳以分析纯化过程中收集的样本,用考马斯 (Coomassie)蛋白染色剂对 PAGE 进行染色 (见图 5)。
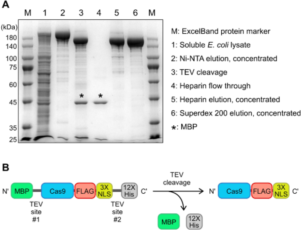
图 5. 重组 Cas9 蛋白的纯度。(A) 在纯化等不同阶段对 Cas9 蛋白进行 SDS-PAGE 电泳分析。(B)重组 Cas9 被表达为融合蛋白,N 端携带一个麦芽糖结合蛋白 (MBP),C 端携带一个 3× FLAG 标签、一个 3× 核定位序列 (NLS)和一个 12× HIS 标签。存在两个 TEV 位点,允许通过 TEV 裂解分离 MBP 和 12× HIS 标签。
B、sgRNA 的合成和纯化
1、sgRNA 的酶法合成
a、通过重叠 PCR 合成 sgRNA 转录模板 (第 1 天)
b、用 T7 RNA 聚合酶进行体外转录 (第 1 天)
2、sgRNA 的 PAGE 纯化
a、变性尿素-PAGE 的浇注 (第 1 天)
b、将 sgRNA 在尿素-PAGE 中进行电泳 (第 2 天)
c、尿素-PAGE 电泳凝胶中 sgRNA 的可视化 (第 2 天)
d、从 PAGE 凝胶中提取 sgRNA (第 2-4 天)
3、去除 5’ 三磷酸基团
a、小牛肠道磷酸酶 (CIP)反应 (第 4 天)
b、sgRNA 的提取和沉淀 (第 4~5 天)
4、sgRNA 地再折叠
a、将 sgRNA 沉淀溶解在 30 μL 的 RNA 溶解缓冲液中,并按照步骤 B-3-a-i 所述确定其摩尔浓度。
b、用 RNA 溶解缓冲液将 sgRNA 浓度调整到 48 μM。
c、将 sgRNA 在 65℃ 下孵育 5 分钟,使 sgRNA 部分展开,然后逐渐冷却到室温以使 sgRNA 重新折叠。
d、将 sgRNA 按 10 μL 等份分装,插入液氮速冻,并储存在 -80℃ 下。sgRNA 可稳定存放2年。应避免反复冷冻和解冻。
C、Cas9 RNP 的组装
- 将 Cas9 蛋白 (40 μM)和 sgRNA (48 μM)解冻至室温。
- 通过混合等体积的 Cas9 蛋白和 sgRNA,建立 20 μM 的 Cas9 RNP 溶液。
- 一边用移液器吸头搅动混合液,一边将 Cas9 蛋白慢慢释放到 sgRNA 中。
- 通过移液管吹吸 4-5 次将溶液混匀,并将 Cas9 RNP 在 37℃ 下孵育 5 分钟,以形成核糖核蛋白复合物。
- 将 Cas9 RNP 保存在室温下并在 2 小时内使用。
D、内毒素检测实验
数据分析
1.用内毒素标准品、凝胶过滤缓冲液、Cas9 和 sgRNA 样本的吸光度读数减去 LAL 试剂水对照品 (空白对照)的吸光度读数。
2.用 Prism 9 软件绘制内毒素标准品的吸光度与内毒素浓度的关系图。
3.确定四个标准浓度之间的最佳拟合直线,得到标准曲线和标准方程 (图 6A)。
4.通过 545 nm 处的吸光度和标准方程 (图 6B)计算凝胶过滤缓冲液、Cas9 和 sgRNA 样本中的内毒素浓度。
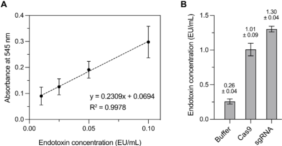
图 6. 分析 Cas9 和 sgRNA 中的内毒素污染。(A)内毒素标准曲线和转换方程。(B)三个重复的凝胶过滤缓冲液、Cas9 和 sgRNA 中内毒素的平均水平。
来源:丁香实验



![考马斯亮蓝G-250 (即用型溶液) [电泳用],阿拉丁](https://img1.dxycdn.com/p/s14/2024/0619/021/4667754395830733081.jpg!wh200)