



包涵体的纯化相关问题

丁香实验
最近在纯化一个GST标签的蛋白,需要用目的蛋白去免疫动物,前期摸条件的时候,发现不管是低温诱导还是37℃诱导,或者是改变IPTG的浓度,破完菌后,目的蛋白在沉淀里面表达都是多很多,我想问一下各位前辈,是破完菌后再用尿素进行溶解破菌沉淀吗?破完菌用尿素溶解后,我直接去与GST进行结合吗(也就是开始纯化了)?我看有说要复性,想问问怎么个前后顺序,复性的话有protocol吗? 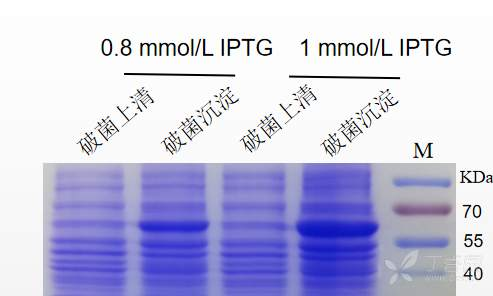
分享

29 个回答

dxyhsx123
有帮助
先破菌,去上清,在洗涤,再用尿素溶解。一些浓度和时间,根据实验进行摸索。

z流沙z
有帮助
首先要破菌好,可以反复冻融一下然后超声裂解,再用尿素进行包涵体溶解,然后对蛋白质进行复性,再进行蛋白质纯化

JCorona
有帮助
可以参考冷泉港的《分子克隆实验指南》,有中译本。里面的参考方法是裂解细菌后离心弃上清,然后摸索尿素的浓度,确定最佳浓度后用含尿素的缓冲液重悬沉淀。溶解后可以直接开始纯化。

lzy必有我师
有帮助
既然决定变复性,就先换个Tag吧,推荐His。 先破菌,然后离心去掉上清,buffer洗一洗,然后再超声一下,再洗洗。最后沉淀留着。 然后加8M Urea/6M GuanHCl +pH(7-8) buffer 去重溶解,再离心去掉不可溶沉淀。 直接上Ni beads 像普通纯化一样,洗脱选用低pH(4-5) buffer。 咪唑没用了。 GST tag做变复性也不是不行,就是变性下tag 结合不了beads。但是His 可以。 洗脱液得到后就可以直接透析复性,最好设置一个Urea梯度透析

内科小护士
有帮助
这个颜色问题好象没有影响吧,只要样品离心了,过滤了,不堵柱子,不是沉淀,上柱没问题,我上次将这种状态的上清过柱子了,结果柱子颜色变褐色了,倒是没堵。但是柱子的结合力下降了。

是小杨同学
有帮助
可以使用尿素溶解,溶解后进行纯化,纯化后就可以复性了

vae1476
有帮助
有试过his tag 吗?GST的tag需要复性后再纯化,his的不需要,建议尝试

ZZDX-YILILI
有帮助
我看图中还是在沉淀中比较多的,也就是最好需要进行包涵体裂解。裂解完之后用尿素进行包涵体溶解。下一步就是对蛋白质进行复性。最好才是利用标签蛋白进行蛋白质纯化。

dxy_rlratyf3
有帮助
可以使用尿素溶解破菌沉淀,不过盐酸胍更优一些。破菌沉淀确实需要复性,但是沉淀里还有很多杂质,需要先用一些洗涤剂比如在50mM Tris,1mM EDTA, pH 7.0-8.5中的2M尿素(过高浓度尿素会让包涵体溶解)除去包涵体上的杂质,再用盐酸胍(6M)溶解,最后复性就是缓慢降低盐酸胍的浓度让蛋白重新折叠,一般稀释法稀释到1.5M盐酸胍的时候复性就结束了。Protocol的话搜索包涵体蛋白稀释复性法就可以了。

verbalkint
有帮助
8M尿素变性沉淀,然后复性,然后才能获得有活性的蛋白去做实验。这里有一个简单的介绍。http://www.friendbio.com/meansMore/id/27

医往情深丁香园
有帮助
破菌不完全不只是指菌体没有破开,还包括菌体虽然已经破开,但是核酸还不够破碎,即形成的核酸片段较大.你所说的这种情况属于后者,造成过大的核酸片段和蛋白结合,同时尿素溶解时会造成出现粘粘的感觉.因此,建议:
1.破菌前先在-20度将菌体冻实,然后再融开破菌.
2.破菌液中加0.1mg/ml的溶菌酶,37度搅半至粘稠,然后粘稠的破菌液变的较稀时再超声,可适当加大超声的功率60HZ应该足够了.
3.超声后,镜检,确保菌体破菌率95%以上.
4.菌体大部分破裂后,加10微克/毫升的DAN酶1,搅拌45分,使大的核酸片段断裂成小片段,然后再离心

一支肾上腺素
有帮助
我觉得是你的蛋白没有变性好才会出现这种情况,更换一下你的变性液,或者改变变性条件试试。

dxy_qkedgrg7
有帮助
使用尿素溶解是可以的,溶解完就可以进行纯化了,然后需要进行复性

dxy_bq4uxnd
有帮助
可以使用尿素溶解,也可以使用一些蛋白保护液溶解,溶解后就可以开始纯化了,使用尿素溶解需要复性,不使用尿素一般不需要复性。

dxy_w4oj7yoe
有帮助
个人觉得可以先尝试优化蛋白可溶性,比如稍微截掉几个氨基酸,或者使用MBP之类更加促溶的tag,也可以尝试昆虫或哺乳动物细胞等更高等的表达体系。为了纯化得到目的蛋白,肯定优先让它表达出可溶蛋白会更好。
如果一定要纯化包涵体,可以使用尿素或盐酸胍溶解,需要复性后才能进行亲和层析,详细纯化方法可以参考以下网站 或者在ncbi上找找相关文献
http://www.detaibio.com/topics/inclusion-body-refolding-introduction.html

jey1235
有帮助
看情况是不可溶的表达。
既然决定变复性,就先换个Tag吧,推荐His。
先破菌,然后离心去掉上清,buffer洗一洗,然后再超声一下,再洗洗。最后沉淀留着。
然后加8M Urea/6M GuanHCl +pH(7-8) buffer 去重溶解,再离心去掉不可溶沉淀。
直接上Ni beads 像普通纯化一样,洗脱选用低pH(4-5) buffer。 咪唑没用了。
GST tag做变复性也不是不行,就是变性下tag 结合不了beads。但是His 可以。
洗脱液得到后就可以直接透析复性,最好设置一个Urea梯度透析。一般就完事了

dxy_h7vcp8v3
有帮助
包涵体的话一般不好纯化。可以尝试其他促溶标签例如MBP,如果,非要纯化。先用加尿素的缓冲液通过超声破碎让蛋白释放出来,然后正常过纯化柱子,正常洗脱,然后洗脱的蛋白,采用透析去掉尿素,然后超滤浓缩复性蛋白

小狐狸FI3I
有帮助
1.如果洗脱组分不纯,可增加洗杂液的体积、调节pH及咪唑浓度、增加一步纯化如离子交换疏水层析等。或对样品进行初步纯化如AS或PEI沉淀后再利用亲和层析完成纯化过程;
2.若洗脱组分中无目的蛋白或蛋白量过低,可通过Western Blotting方法对His标签蛋白表达和纯化样品进行定性和定量分析。如果蛋白表达量过低,则可尝试改变和优化表达条件来提高产量;如果蛋白表达量正常但是纯化效果不佳,则可尝试降低洗杂液浓度、提高洗脱液浓度以防止结合较弱或过强的目的蛋白的损失;
3.倘若上样过程中出现蛋白沉淀现象,则可能是操作温度太低,可以待样品恢复至室温后上样。也可考虑在样品或缓冲溶液中添加稳定剂。

zxb597
有帮助
破菌不完全不只是指菌体没有破开,还包括菌体虽然已经破开,但是核酸还不够破碎,即形成的核酸片段较大,洗涤之后的包涵体沉淀最好不要直接加8M尿素溶解,那样很容易在包涵体表面形成透明不溶物。
隔壁家的小夜猫子
有帮助
对的,用尿素裂破菌后的沉淀,直接可以结合亲和层析,亲和之后透析复性


相关产品推荐




相关问答
提问
扫一扫

实验小助手

扫码领资料
反馈
TOP
打开小程序