RNA测序简介
北京启衡星生物科技有限公司
NGS已成为生命科学领域中基因组学和转录组学研究的金标准。虽然它与桑格测序等方法有某些核心相似之处,但NGS所能实现的绝对的高通量水平使它与其他方法截然不同,并彻底改变了科学家的工作方式。
RNA测序(RNA-seq)尤其处于NGS功能应用的前言沿。RNA-seq最简单的形式允许我们在取样时确定样本中的RNAs的丰度。转录组是一个高度动态的细胞特征,并打开了一个巨大发掘潜力的世界。对药物、各种疾病状态、转录后修饰和选择性剪接转录本的反应变化只是 RNA-Seq 使其能够发现的一些例子。
简而言之,RNA-Seq 使我们能够以前所未有的灵敏度评估整个转录组,并在特定于所研究主题的条件和时间点下生成转录组的快照。尽管这是对更广泛的 RNA-Seq 领域的非常合适和简短的总结,但仍有许多类型的 RNA-Seq 可供尝试。
短读长测序是 RNA-seq 的常用方式。测序通常从样本材料中生成的较短的插入片段,长度在 50 – 500 bp 之间,然后在下游数据分析中重新组装或计数。长读长测序经常在 DNA-Seq 和 RNA-Seq 中以高容量确定手头样本的序列,例如,一次 10,000 – 100,000 个碱基对。对于 RNA 测序,结合长读长和短读长测序对于确定未知或未注释物种的转录组特别有用。长读长测序可以确定准确的转录本序列,并提供一个支架,短读长可以像拼图一样组装在该支架上。
Bulk RNA-Seq 测量的是一组样品的基因表达,而不区分样品中的细胞类型。该方法提供了一组样本中基因表达的广泛概述。RNA-Seq 应用的范围极其广泛,而且界限日新月异(参见下面的列表示例应用)。有了如此广泛的应用,毫不为奇RNA-Seq是一种有用的、强大的工具,可以提供一系列实验样本的定量、定性和时间分辨数据。
常规RNA-Seq应用场景
-
alternative polyadenylation events(可变腺苷酸化位点分析),
-
alternative splicing analysis(可变剪切分析),
-
biomarker discovery(生物标记物发现),
-
cell type and subtype characterization(细胞类型及亚型表征),
-
differential expression(差异表达分析),
-
discovery of novel genes and gene isoforms(新基因和基因亚型的发现),
-
expression quantification(基因表达定量),
-
host / pathogen interaction (dual RNA-seq)(宿主/病原体相互作用),
-
identification of genuine transcription start sites (and promoters)(真正转录起始位点(和启动子)的鉴定),
-
metatranscriptomics (community transcriptomes)(宏转录组分析),
-
precise 3’ end mapping(3‘端精确mapping),
-
RNA kinetics (biosynthesis and decay)(RNA动力学分析(生物合成和降解)),
-
tissue-specific gene expression(组织特异性基因表达分析),
-
transcriptome assembly of uncharacterized species,
and many other applications(未知物种的转录组组装和其他应用).
-
从样本中分离出RNA,并除去污染的DNA,例如,用DNase处理。 -
如果需要,则对RNA进行预处理,即通过polyA选择或rRNA去除来富集mRNA。 -
RNA片段化(如果使用无片段化建库流程如Corall total RNA seq library preparation kit,这一点可以忽略)。 -
利用所选的RNA样本和文库生成引物进行逆转录进行一链合成。 -
然后通过沿cDNA链随机引物进行第二链合成。 -
进行末端修复,接头连接。 -
最后进行PCR扩增,为文库添加index,或文库扩增以以获得足够量的文库,以及进行文库质量控制。
Lexogen mRNA-Seq文库准备与其他全转录组测序文库制备方法略微不同。在Lexogen mRNA-Seq文库准备中,接头部分序列已经在第一步引入,例如在反转录过程中。因此,通过删除各种处理步骤,使工作流程得以简化和有效地缩短时间。允许在几个小时内完成文库的准备工作流程。图1展示了两种不同RNA-Seq文库制备方法的工作流程示意图。
RNA-Seq 可用于多种应用场景,可通过不同方法的组合高度定制,并提供无限的修改可能性,以满足研究项目的个性化需求。在拿起移液器之前计划好你的实验是成功的关键,确保高质量的结果来支撑你的想法和假设。
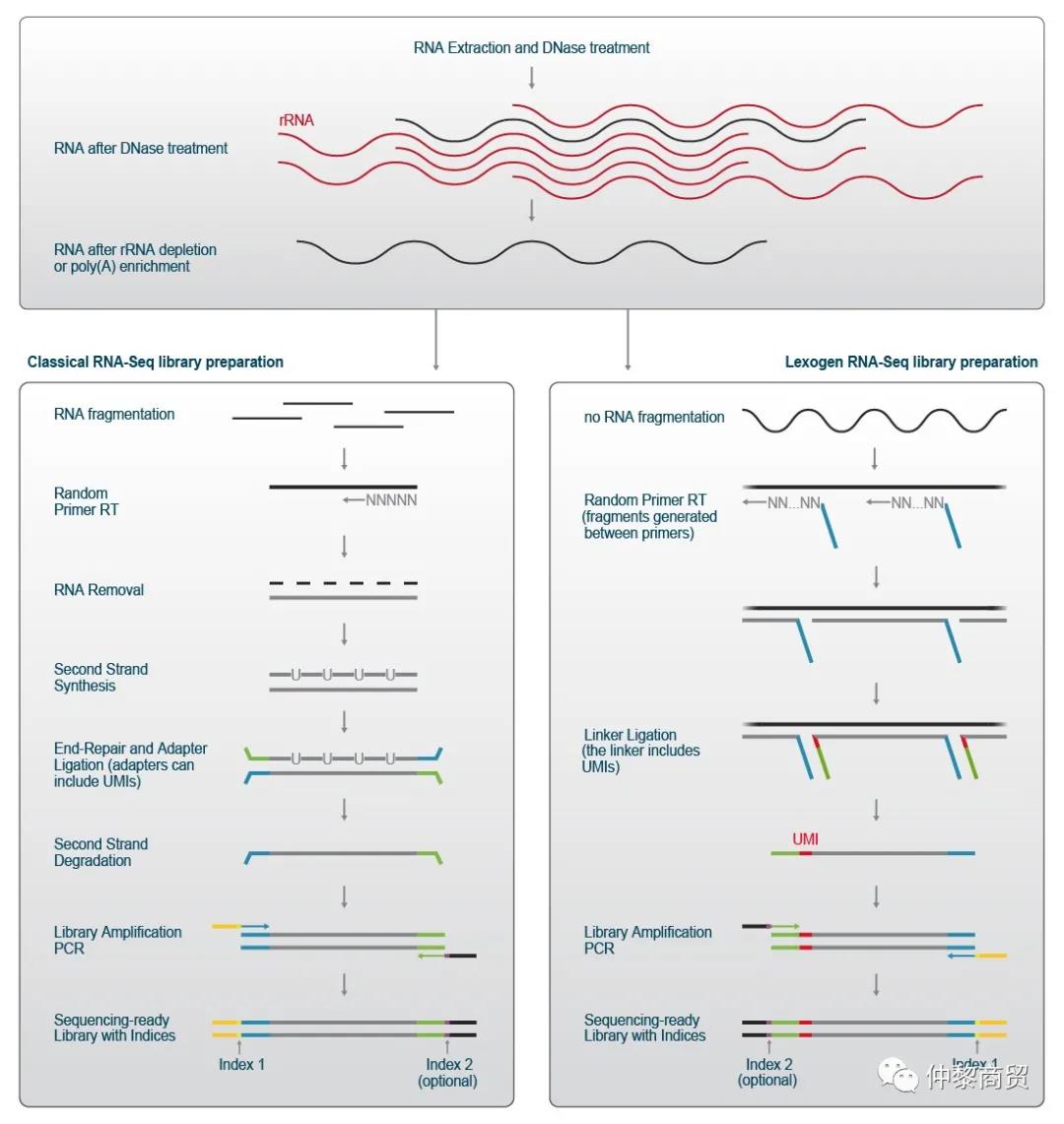
图1 Illumina短读长测序| RNA-Seq文库准备流程RNA从细胞、组织、生物液体或其他样本中提取,并在核糖体RNA去除或多聚(A) RNA富集之前,用DNase去除基因组DNA。左:描绘经典 RNA-Seq 工作流程的示意图概述。预处理的 RNA 被片段化,cDNA 由随机引物逆转录产生。去除 RNA 后,产生第二条链。用 dUTP 标记允许生成保留链的文库。双链 cDNA 被末端修复,测序接头被连接,dUTP 标记的链被特异性降解。从而,仅保留一个链,并且保留链信息。对该剩余链进行 PCR 以扩增文库、完成用于测序的接头序列并引入index。右图:Lexogen RNA-Seq 文库制备方案省略了 RNA 片段化,cDNA 第一条链是从已经包含部分接头序列的随机引物中生成的。在两个杂交的引物之间产生插入片段,第二个部分接头连接到 cDNA 的第一条链,从而保持链特异性。 接头还包含单分子标签 (UMI),将在单独的章节中讨论。然后通过 PCR 获得包含 UMI、全长接头和index的测序文库。