做PCR达人,从引物设计开始
普健生物(武汉)科技有限公司
引|物|设|计
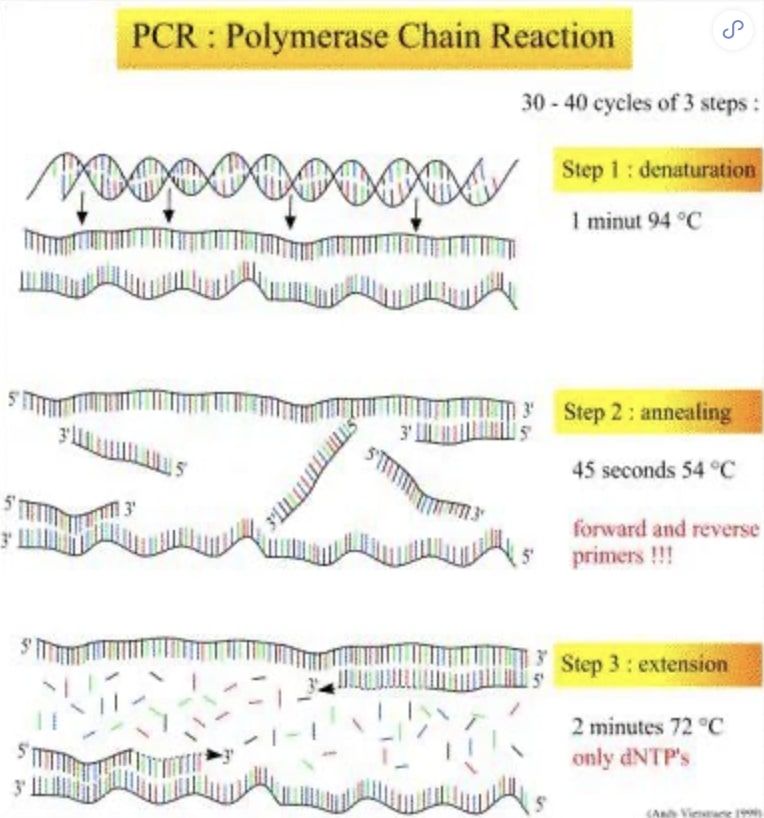
20世纪后期发展的PCR技术改变了整个生物学研究的进程,其中引物设计的好坏直接关系到PCR的成败。
引物设计的基本原则
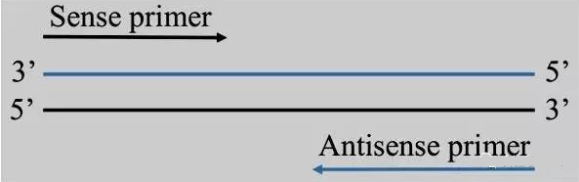
引物设计的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。在某些情况下(比如构建文库)是在不知道模板序列的情况下进行引物设计的,这个时候引物核苷酸序列与模板不是完全匹配。我们通常的引物设计都是在已知模板序列的情况下进行,如下图1所示:
▲
图1 引物设计示意图
引物设计总体上包含三个程序:模板的获得、同源性分析以及引物筛选。模板的获得有多种来源,根据实验目的来定,这里不多作讨论。同源性分析的目的是看引物与模板有没有非特异性结合位点,特别要避免非特异性结合位点的3’末端完全配对。为最大程度确保获得较好的PCR结果,引物筛选需要满足以下几项基本原则:
01
引物长度:18-30 nt
大多数应用的最短引物长度为18个核苷酸,引物长度的上限并不是很重要,主要与反应效率有关。引物越长,退火结合到靶DNA上形成稳定双链模板的速率越小。特殊情况下可以进一步延长到50-60 bp,但长引物的合成出错率高,价格贵,而且难以合成,因此较为少见。另外,上下游引物的长度最好基本相等,最多相差不超过3 bp。
02
引物GC含量:40-60%
A、T、G和C四种碱基要在引物中均匀分配,如果引物存在严重的GC倾向或AT倾向则可以在引物5’端加适量的A、T或G、C尾巴。
03
引物的退火温度:55~75 ℃
退火温度需要比解链温度(Tm)低5 ℃,如果引物碱基数较少,可以适当提高退火温度,反之亦然。一对引物的退火温度相差4~6 ℃不会影响PCR的产率,但是理想情况下一对引物的退火温度是一样的。扩增产物的Tm值与引物的 Tm值相差不能大于10 ℃。
04
避免扩增模板的二级结构区域
用有关计算机软件可以预测目的片段的稳定二级结构,选择扩增片段时最好避开模板的二级结构区域。
05
引物末端不应超过3个连续的G或C
引物在G+C富集区容易错误引发,除在特殊的PCR(AS-PCR)反应中,引物3’端不能发生错配。如扩增编码区域,引物3’端不能终止于密码子的第3位,因为密码子的第3位易发生简并,会影响扩增特异性与效率。
06
引物不能形成二级结构
引物自身存在的互补序列会折叠成发夹结构从而影响其与模板的结合。若用人工判断,引物自身连续互补碱基不能大于3 bp。两引物之间也不应该存在互补性,尤其应避免3’端的互补重叠以防止引物二聚体的形成。一般情况下,一对引物间不应多于4个连续碱基的同源性或互补性。
07
为了后续操作而产生的不完全匹配
5’端对扩增特异性影响不大,因此可以被修饰而不影响扩增特异性,这些修饰包括:添加酶切位点、添加标记(生物素、荧光、地高辛和Eu3+等)、引入与蛋白质结合的DNA序列、引入突变位点、插入与缺失突变序列和引入启动子序列等。
特定PCR的引物设计
在尽量遵循引物设计基本原则的同时,根据不同的实验目的,需要注意一些相应的事项,总结如下:
01
荧光定量PCR
荧光定量PCR有染料法和探针法两种。染料法只需要设计引物,而探针法除设计引物之外还得设计一条探针。
引物设计要尽量满足以下要求:
确保模板是cDNA而不是gDNA;避免重复碱基(尤其是G);GC含量为30-80%;Tm值是58-60 ℃;3’端最后5个碱基内不能有超过两个的G或C;产物长度一般为80-150 bp最为合适;引物应尽可能跨外显子以免受基因组DNA的影响;查看有无假基因(无功能的DNA序列,与需要扩增的目的片段长度相似)存在。
TaqMan探针设计应注意:
探针位置尽可能靠近扩增引物,但不能与引物重叠;长度一般为18~40 bp;GC含量控制在40~80%左右;避免连续相同碱基的出现,特别是要避免GGGG或更多G出现;在引物的5’端避免使用G;选用比较多的碱基C;退火温度控制在68~70 ℃左右。
02
多重PCR
由于多重PCR体系中同时存在多对引物,在引物设计时应注意:各引物对必须保持高度的特异性,避免非特异性扩增;尽可能避免所有引物间的相互作用;各引物对应保持相对一致的扩增效率,且不同引物对扩增出来的产物能通过电泳或其他方法区分开。
03
Nest-PCR
Nest-PCR,也叫巢式PCR,是一种变异的PCR,使用两对而非一对PCR引物扩增完整的目的基因片段。第一对PCR引物扩增的片段与片段准相近,第二对引物称为巢式引物,与第一段PCR产物结合来指导第二段PCR产物的合成,这段产物比第一段产物要短。巢式PCR的好处是如果错误的PCR产物被扩增,那么由第二段引物指导的第二次扩增的产物错误的可能性将变得非常低。
04
RACE
RACE(Rapid-amplification of cDNA Ends)是基于PCR技术基础上由已知的一段cDNA片段,通过往两端延伸扩增从而获得完整的3’端和5’端的方法。一般分5’ RACE和3’ RACE两种。
3’ RACE较简单,首先将mRNA或总RNA用Poly T引物反转录,根据一般基因具有polyA尾巴的特点,选用特异引物(根据已知序列设计)和Poly T引物PCR即可。
5’ RACE相对较难,目前流行几种5’ RACE。其一为加接头(传统),根据接头引物和自己设计特异引物PCR,可以设计巢式PCR二次扩增。另外,有利用反向PCR技术,连接成环再PCR。
特异性引物应该满足:
长度为23~28 nt;50~70%的GC含量;Tm值≥65 ℃;避免使用与AP1互补的引物,尤其是在3’末端;如果要用重叠片段来检测设计的引物,GSp1和GSp2之间至少是100~200 bp。
05
测序
一般来说,测序引物的设计要比普通PCR严格,应遵循以下原则:长度15~25 bp,一般常用20 bp(根据GC含量作适当调整);3’端应尽量选择G或C碱基;Tm值为50~70 ℃;GC含量在50%左右,尽量避开A、T、G、C的连续结构;避开引物自身形成发夹结构或引物二聚体结构等复杂结构;保证引物和模板100%匹配,特别是3’端的几个碱基一定要完全匹配,同时必须严格保证引物和模板之间只能有一个结合位点。
06
简并引物的设计
简并引物是根据某些特定的目的而选用的一组混合物,指在寡核甘酸的某一位置(一个简并位)上有多个碱基存在于不同的寡核甘酸分子中。
简并引物常用于从已知蛋白到相关核酸分子的研究以及用一组引物扩增一类分子。在上述两个主要应用中,需要注意以下两个问题:
1从蛋白到核酸,应注意:尽量选择简并度低的氨基酸区域为引物设计区;充分注意物种对于密码子的偏好性,选择该物种使用频率高的密码子,以降低引物的简并性;引物不要终止于简并碱基,对于大多数氨基酸残基来说,意味着引物3’末端不要位于密码子的第三位;在简并度高的位置,可用次黄嘌呤(dI)代替简并碱基。
2用一对简并引物扩增一类DNA分子时,同样遵循上述总的原则,即尽量降低引物的简并度,尤其在3’末端或近3’末端。 在此种应用中,应先利用工具软件或其他工具找到这些分子的保守区,然后根据共有序列,应用上面所述的一些原则来设计所需要的引物。
常用的软件
软件的引物设计功能主要体现在两个方面:首先是引物分析评价功能,该功能只有少数商业版软件能够做到,其中以“Oligo 6”最优秀;其次是引物的自动搜索功能,各种软件在这方面的侧重点不同,因此自动搜索的结果也不尽相同。自动搜索功能以“Premier Primer”最强且方便使用,“Oligo 6”其次,其他软件如"Vector NTI Suit"、“Dnasis”、“Omiga”和“Dnastar”都带有引物自动搜索功能,但搜索结果不是十分理想。要想得到效果很好的引物,在自动搜索的基础上还要辅以人工分析。引物设计软件的最佳搭配是“Oligo”和“Premier”软件合并使用,以“Premier”进行自动搜索,“Oligo”进行分析评价,如此可快速设计出成功率很高的引物。
然而,许多引物设计的基本原则在实际应用中往往难以做到都符合,我们只能秉着“实践是检验真理的唯一标准”这一原则,通过实验验证是否可行,再根据结果做出调整。
最后, AtaGenix
祝各位实验达人一切顺利!