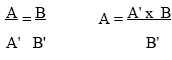Quantitative PCR
互联网
ABSTRACT
Quantitative PCR involves co-amplification of two templates: a constant amount of a preparation containing the desired target sequence and varying amounts of a reference template. After amplification, the concentration of the target sequence in the preparation of nucleic acid under test is established by interpolation into a standard curve. Quantitation of nucleic acids by PCR is best performed by real-time PCR. However, the following robust protocol, which uses radioactivity to quantify PCRproducts, remains useful when a real-time instrument is unavailable. The method can be easily adapted to other methods of quantification such as fluorometry.
MATERIALS
Reagents and Solutions
Chloroform dNTP solution (pH 8.0), containing all four deoxynucleotide triphosphates, each at a concentration of 20 mM MgCl2 (1 M)
Placental RNase inhibitor (20 units/µl)
Enzymes and Buffers
Appropriate restriction enzymes and 10x buffers
Bacteriophage T4 DNA ligase and 10x buffer
Reverse transcriptase, required only if RNA is used as a template
Thermostable DNA polymerase and 10x amplification buffer as supplied by the manufacturer or homemade
500 mM KCl
100 mM Tris-Cl (pH 8.3, room temperature)
15 mM MgCl2
Nucleic Acids/Oligonucleotides
DNAmarkers for gel electrophoresis
Externally added reference (either DNA or RNA) of known concentration
Use a DNA reference to measure the concentration of DNA sequences and, if possible, an RNA reference for RNA targets. A method to construct reference RNA is described in Protocol 15.2.
Sense and antisense primers, each 20 M in H2O
There is nothing unusual about the primers used in quantitative PCR. The standard rules for primer design apply.
Target nucleic acid
The target can be a preparation of DNA or RNA, either total or poly(A)+. Dissolve preparations of total RNA in H2O at a concentration of 0.5-1.0 mg/ml and preparations of poly(A)+ RNA at 10-100 µg/ml. Dissolve DNA targets in 10 mM Tris-Cl (pH 7.6) at the following concentrations:mammalian genomic DNA, 100 µg/ml; yeast genomic DNA, 1 µg/ml; bacterial genomic DNA, 0.1 µg/ml; and plasmid DNA, 1-5 ng/ml.
Radiolabeled Compounds
[α-32]dCTP (sp. act. 3000 Ci/mmole at 10 mCi/ml)
Gels/Loading Buffers
Polyacrylamide or agarose gel
Additional Items
Barrier tips for automatic pipettor
Fluorometer (optional; see Step 1)
Light mineral oil or wax bead (optional; see Step 5)
Materials for autoradiography or phosphorimaging
Microtiter plates or microfuge tubes, 0.5 ml and thin walled
Positive displacement pipette
Thermal cycler, programmed with desired amplification protocol
Water baths (94℃and, for RNA templates only, 75℃)
METHOD
Design and prepare a reference template suitable for the task at hand. Measure the concentration of the reference template as carefully as possible, preferably by fluorometry. Alternatively, estimate the amount of reference template after gel electrophoresis and ethidium bromide staining.
Make a series of tenfold dilutions (in H2O) containing concentrations of the reference template ranging from 10-6 to 10-12 M. After using the dilutions (Step 3), they should be stored at -70℃ for later use in Step 8.
If starting from RNA, denature the target RNA by incubating aliquots for 5 minutes at 75°C, followed by rapid chilling in ice water. Then, without delay, set up a series of reverse transcription reactions containing increasing amounts of reference template in sterile 0.5-ml microfuge tubes. For each reaction in the series, prepare the following:
10x amplification buffer 2 µl
20 mM solution of four dNTPs (pH 8.0) 1 µl
20 µM antisense primer 2.5 µl
approximately 20 units/ µl placental RNase inhibitor 1 µl
50 mM MgCl2 1 µl
denatured target RNA 10 pg to 1.0 µg
100-200 units/ µl reverse transcriptase 1 µl
tenfold dilution of reference template 1 µl
H2O to 20 µl
Incubate the reaction for 60 minutes at 37℃ and then denature the reverse transcriptase by heating to 95℃ for 20 minutes.
In sterile 0.5-ml microfuge tubes, amplification tubes, or the wells of a sterile microtiter plate, set up amplification reactions with each reaction in the series from Step 3:
everse transcriptase reaction (Step 3) or target DNA 1 µl
20 µlM sense primer 1.5 µl
20 µlM antisense primer 1.25 µl
10x amplification buffer 5 µl
[α-32P]dCTP (3000 Ci/mmole) 10 µCi
20 mM solution of four dNTPs 1 µl
thermostable DNA polymerase 2 units
H2O to 50 µl
IMPORTANT:
Do not reduce the concentration of unlabeled dCTP in the reaction mixture to increase the specific activity of the precursor pool. There is a danger that the amount of the nucleotide could become limiting at late stages in the amplification reaction.
If the thermal cycler is not fitted with a heated lid, overlay the reaction mixtures with 1 drop (~50 µl) of light mineral oil. Alternatively, place a bead of wax into the tube if using hot start PCR. Place the tubes or the microtiter plate in the thermal cycler.
Amplify the nucleic acids using the denaturation, annealing, and polymerization times and temperatures listed in the table.
Cycle Number Denaturation Polymerization Annealing
30 cycles 30 sec at 95℃30 sec at 55℃1 min at 72℃ Last cycle 1 min at 94℃ 30 sec at 55°C 1 min at 72℃
Times and temperatures may need to be adapted to suit the particular reaction conditions.
When using a reference template that differs from the target sequence in size:
Analyze and quantitate the amplified products.
Analyze the sizes of the amplified products in a 20- µl aliquot of each of the reactions by gel electrophoresis and autoradiography.
Excise the amplified bands of the control template and target sequences from the gel and measure the amount of radioactivity in each band in a liquid scintillation counter. Alternatively, scan the gel with the appropriate detector (e.g., GEHealthcare scanner or phosphorimager).
Calculate the relative amounts of the two radiolabeled DNAs in each of the PCRs.
Correct the amount of radioactivity to allow for differences in the molecular weights of the two radiolabeled DNAs
When using a reference template that contains a novel restriction site or lacks a naturally occurring site.
Heat the samples to 94°C for 5 minutes following the final round of amplification.
Allow the samples to cool gradually to room temperature and then digest a 20-µl aliquot of each of the reactions with the appropriate restriction enzyme .
Analyze the sizes of the amplified DNA fragments by gel electrophoresis and autoradiography or phosphorimaging.
Excise the amplified bands of the control template and target sequences from the gel and measure the amount of radioactivity in each band in a liquid scintillation counter. Alternatively, scan the gel with the appropriate detector (e.g., GEHealthcare scanner or phosphorimager).
Calculate the relative amounts of the two radiolabeled DNAs in each of the PCRs.
Correct the amount of radioactivity to allow for differences in the molecular weights of the two radiolabeled DNAs.
Examine the results to determine the concentration of reference template that yields approximately the same amount of amplified product as the target sequence. Set up a second series of amplification reactions (please see Step 4) containing a narrower range of concentrations of reference template.
It is best to generate this series of dilutions from the appropriate tenfold dilution of the reference template (Step 2).
Repeat Steps 5-7. For each amplification reaction, measure the ratio of the yield of amplified reference template to the yield of amplified target sequence. Plot this ratio against the amount of reference template added to each amplification reaction. From the resulting straight line, determine the equivalence point (i.e., the amount of reference template that gives exactly the same quantity of amplified product as the target sequence in the reaction). Calculate the concentration of the target sequence in the original sample.
Anyone using the procedures in this protocol does so at their own risk. Cold Spring Harbor Laboratory makes no representations or warranties with respect to the material set forth in this protocol and has no liability in connection with the use of these materials. Materials used in this protocol may be considered hazardous and should be used with caution. For a full listing of cautions regarding these material, please consult:.
Molecular Cloning: A Laboratory Manual Third Edition, by Joseph Sambrook and David W. Russell, © 2001 by Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, p. 8.86-8.95.
Copyright © 2006 by Cold Spring Harbor Laboratory Press. All rights reserved. No part of these pages, either text or image may be used for any purpose other than personal use. Therefore, reproduction modification, storage in a retrieval system or retransmission, in any form or by any means, electronic, mechanical, or otherwise, for reasons other than personal use, is strictly prohibited without prior written permission.