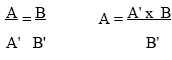定量RT-PCR (Quantitative RT-PCR)
互联网
Application: Quantitative RT-PCR is used to quantify mRNA in both relative and absolute terms. It can be applied for the quantification of mRNA expressed from endogenous genes, and transfected genes of either stable or transient transfection. It is the most sensitive method as yet in quantitative analysis of mRNA. Principle: PCR amplification follows the formula:
A = B(1 e)n
A=amplified products, B=input templates, n=cycle number, and e=amplification efficiency. Factors affecting amplification efficiency in the RT-PCR process include the efficiency of reverse transcription, Mg 2 / dNTPs/ primer concentrations, enzyme activity, pH, annealing temperature, cycle number, temperature variation, tube to tube variation etc. Since PCR results in a million fold amplification, variation in any of the above factors during the amplification process will significantly affect the final output; therefore routine RT-PCR can not be used for the purpose of quantitative analysis. This elimination, however, can be overcome using quantitative RT-PCR (1). This method uses an external template as the internal control for all the steps in RT-PCR process. The quantitative RT-PCR follows the formula:
A = B (1 e)n
A' B'(1 e)n
A=amplified products, B= input templates, A'=amplified control products, and B'=input control templates. Any effect on the amplification efficiency will equally affect both templates, thus providing a linear relationship between both wild type and control templates in the amplification process. The output ratio between the two templates directly reflects the input ratio between these two templates.
As the amount of input control templates is known, the amount of wild type templates in the RNA sample can be easily determined.
The system described below has been developed for the quantification of endogenous expression from either single or multiple genes. The expression from several genes can be simultaneously quantified in both relative and absolute terms. In comparison to other RNA quantification methods, this assay is highly sensitive, highly quantitative, and gives cleaner results when performed properly. RT-PCR can be used for analyzing expression from single or multiple genes, and analyzing changing expression patterns in diseases etc.
Procedure:
1. RNA Preparation
A number of procedures are available for preparation of RNA from tissue culture cells or mononuclear cells. The NP-40 lysis method affords a good choice (5) for preparation of RNA from tissues or primary cells, however, guanidine method is better(6). From our experience, a commercial product Trizol solution (GIBCOBRL), a modified guanidine solution, gives the best result. For isolation of RNA from white blood cells, first use PMN solution (Robbions Scientific Corporation) to isolate white blood cells which will include all mononuclear and polymorphonuclear cells. The isolated RNA should be treated with DNase I to remove genomic DNA contamination. Quantify the RNA by measuring O.D260 and check the quality on an agarose gel. Typically, the RNA yield from a 20 ml blood sample is about 20 mg.
Grow cells in exponential phase. If desired, activate the gene with conditions such as heat shock, etc. Prepare RNA for the analysis of endogenous gene expression or for the analysis of a stable transfected gene (for studying the expression of a transient transfected gene, you will need to co-transfect the wild type plasmid with a control plasmid containing the modified homologous gene controlled by a constitutive promoter, in order to control the transfection efficiency (2, 3).
During RNA isolation and analysis, it is essential to maintain an RNase free environment. All solutions should be RNase free. Always wear gloves. Keep samples on ice and immediately freeze RNA samples after using them (4).
The following procedure is a modified version of the NP-40 RNA extraction method for tissue culture cell lines (5). It is simple, fast and gives high yield of high quality RNA.
RNA can be obtained in less than one hour.
a. Spin down 0.1-1 x 107 cells.
b. Resuspend cells in 200 ml cold buffer A:
Buffer A:
10 mM HEPES pH 7.9
10 mM KCl
0.1 mM EDTA
0.1 mM EGTA
2.5 mM DTT
c. Add 10 ml of 10% NP-40, vigorously vortex for 10 seconds.
d. Spin 30 seconds, transfer 200 ml supernatant to a new tube.
e. Add 200 ml of buffer B at room temperature, 400 ml chloroform-phenol,vortex 1 minute.
Buffer B:
7 M Urea
1% SDS
0.35 M NaCl
10 mM EDTA
10 mM Tris pH 7.5
f. Centrifuge for 10 minutes at 13K RPM in microcentrifuge.
g. Recover 360 ml upper phase, add 900 ml 100% EtOH, mix, spin for 15 minutes at room temperature.
h. Wash pellet with 70% EtOH, dry and dissolve the pellet in 10-50 ml H2 O.
i. Quantify RNA yield from OD260 value. Average yield is about 5 mg per
106 cells.
j. Check RNA quality by loading 1 ml of RNA on a 1.5% agarose gel. Two ribosomal RNA bands should be intact to ensure no degradation occurred.Any degraded RNA samples should not be used for the quantitative analysis.
2. Primer Design for RT-PCR
a. Design both 5' sense primer and 3' antisense primer for PCR following the classical parameters for primer design. The 3' primer will also be used for reverse transcription. The length of the amplified fragment should be within a reasonable range, e.g., around 200-600 bp, for the preparation of control template, later fragmentation and quantitative analysis.
b. Calculate the Tm for both primers with the formula:
Tm (0 C) = 2 (A T) 4 (G C)
c. Perform PCR with the primers and plasmid containing the wild type templates. If there is difficulty in the amplification, it may be very helpful to test different [Mg2 ]. Titrate the [Mg2 ].from 0.5 mM to 5 mM and run
the PCR to determine the optimal [Mg2 ].
3. Preparation of Internal Control RNA Template
The aim is to make a small internal deletion in the cDNA between the two primers used for PCR . The modified template will be used to make a control product which is shorter than the product obtained with wild type RNA.
a. Clone the cDNA fragment containing the region between the 5' and 3' primers into a suitable vector which has SP6, T3, or T7 promoters.
b. Find suitable restriction sites within the cloned fragment, perform a restriction digestion and re-ligate both ends. The deleted portion should be a size which ensures minimum difference between the wild type and shorter control templates while making it easy to separate these two templates later.
c. Purify and sequence deleted plasmid DNA construct to confirm the deletion is correct.
d. Perform a 10 x mini-preparation of the plasmid DNA .
e. Linearize the plasmid with the deletion by restriction digestion. The site chosen should be downstream of the 3' antisense primer. The end created should be either 5' protruding or blunt. Blunt any 3'protruding end with Klenow.
f. Purify the linearized plasmid with Geneclean beads and dissolve the DNA in DEPC treated H2 O.
g. Incubate the purified DNA with 1 mg RNA at 37°C for 1 hour and load the mixture on a 1.5% agarose gel to check RNA quality. Any degradation of RNA will indicate the presence of RNase activity in the plasmid DNA preparation. Re-purify the DNA and repeat the steps above to ensure that no RNase activity remains.
h. In- vitro Transcript Preparation (also see reference 6):
i. Add the components at room temperature in the following order:
ii. Incubate 2 hours at 40°C for SP6 polymerase, or at 37°C for T3 and T7 polymerases.
iii. Load 1 ml on an agarose gel to check the synthesis of the transcripts, comparing with template DNA alone.
iv. Add 2 ml RNase free DNase I (Phamarcia, 10 u/ml), vortex thoroughly, spin briefly, incubate at 37C for 1h to destroy DNA templates.
v. Add 100 ml chloroform-phenol, vortex, centrifuge, recover 100 ml upper aqueous phase.
vi. To remove the free nucleotides, add 50 ml 7.5 M NH4OAC, 200 ml 100% EtOH, keep at -70°C for one hour, spin at 4°C, remove the supernatant.Dissolve the pellet in 100 ml 1 M NH4OAC, add 200 ml 100% EtOH, keep -70°C for 1 hour; spin, wash the pellet with 70% EtOH, and dissolve the pellet in 20 ml H2 O.
vii. Quantify the transcripts at OD260 and check the quality on an agarose gel.
viii. Carefully make a series of 10x dilutions in a 200 ml volume from 10 ng/ml to 10 fg/ml, vortex each one and change tips before making next dilution. Store the samples at -70°C. These will be used as control RNA.
4. RNA Purity Testing
Add 1 mg/ml DNase free RNase A to 1 mg/ml RNA, and 10 ng/ml control RNA. Incubate at 37°C for one hour. Perform PCR to test if there is any amplification. Any amplification will indicate the presence of a genomic copy of the wild type gene in the RNA sample preparation and plasmid template contamination in control RNA preparations. Use DNase I digestion to remove these DNA templates. Check again to ensure the purity of sample and control RNA preparations.
5. Determination of Parallel Range Between Wild Type Templates and Control
Templates For multiple analyses, the parallel range for each gene needs to be determined.
a. Prepare RNA master mix. Add 10 mg of RNA in 75 ml H2O, mix well, aliquot to 10 wells in a PCR plate (8.5 ml per well =1 mg per well).
b. Add 1 ml of the diluted control RNA in each well in the order of 10 ng, 1 ng, 100 pg, 10 pg, 1 pg, 100 fg, 10 fg, 1 fg. c. Add 10.5 ml reverse transcription mixture to each well.
RT mixture:
3' antisense primer (20 pmol/ml) 1 ml
optimized concentration of MgCl 2
10 x PCR buffer (500 mM KCl, 100 mM Tris-HCl, pH 8.3) 2 ml
dNTP mixture (5 mM each) 2 ml
DTT (100 mM) 2 ml
RNase inhibitor (35 u/ml) 0.1 ml
MMLV reverse transcriptase (Phamarcia, 14 u/ml) 0.2 ml
H2 O to 9.5 ml
d. Incubate the plate at 37°C for 1 hour.
e. Add 30 ml of PCR mixture in each well and overlay with 2 drops of paraffin oil
PCR mixture:
f. Run PCR using the conditions of 94°C for 2 minutes, then 25 to 35 cycles of 94°C-1 minute (depends on the abundance of target templates) followed by the annealing temperature (2 degrees lower than calculated Tm) for 1 minute followed by 1 minute of extension at 72°C.
g. Run 8 ml of RT-PCR products on a 2% agarose gel. There will be a size shift between the wild type template and the control template bands caused by the deletion. This is where the amounts of input wild type template and control template are at parallel levels.
6. RT-PCR Co-Amplification of Wild Type and Control Templates Using the amounts of control RNA defined above, prepare a master mixture of control RNA in H2 O, aliquot to each well, and add 1 mg/ml of total RNA. Perform RT-PCR as above with the addition of 0.1 ml of [a-32 P] dATP in PCR mixture to label the amplified products. Set positive control with RNA alone, control RNA alone, and negative control without input of any templates.
7. Fractionation of Amplified Wild Type and Control Templates
a. Load 5 ml of RT-PCR sample on 2% agarose gel.
b. According to the signal intensities in agarose gel, determine the relative amount of different samples to be used for fragmentation to obtain similar intensities for different samples. Usually an average of 5 ml should be enough to give significant signals.
c. Remove a defined amount from the different samples and vacuum dry the sample.
d. Thoroughly wash the dried sample with 70% EtOH. Spin 5', remove supernatant, vacuum-dry the pellet, dissolve in 3 ml sequence gel loading buffer, boil and load on a sequencing gel of suitable concentration (from 4% to 8%) to separate the amplicons. Dry the gel and expose it to a PhosphorImager plate.
8. Determination of the Signal Values of Amplified Wild Type and Control Templates. Use the PhosphorImager to scan the signals from both wild type templates and control templates. Subtract the background.
9. Calculation of the Amount of Input Wild Type Templates. a. Calculate the ratio between the wild type templates and the control templates: Ratio between the wild type templates and the control templates=
signal value of wild type amplicons
signal value of control amplicons
This value can be used directly for the relative comparison of gene expression under different conditions.
At this point, you can also calculate the absolute amount of input wild type mRNA, the total number of molecules in the input wild type mRNA, and the total number of molecules of the wild type mRNA per cell (see references).
References:
1. Gilliland G., S. Perring, K. Blanchard, and H. F. Bunn: Analysis of cytokine mRNA and DNA : Detection and quantitation by competitive polymerase chain reaction. PNAS 87:2725-2729, 1990
2. Sperisen P., S. M. WANG, P. Reichenbach, and M. Nabholz: A PCR -based Assay for Reporter Gene Expression. PCR Methods and Application 1: 164-170, 1992
3. Sperisen P., Wang S. M., E. Soldani, M. Pla, C. Rusterholz, P. Bucher, P. Reichenbach, and M. Nabholz. Mouse Interleukin-2 Receptor a Gene Expression. JBC 270(18):10743- 10753, 1995
4. Sambrook J., E.F. Fritsch, and T. Maniatis: Chapter 7. Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), 1989
5. Gough N. M.: Rapid and quantitative preparation of cytoplasmic RNA from small numbers of cells. Anal. Biochem. 173: 93-95, 1988
6. Promega: Protocols and Applications Guide: 2nd edition. p58-61, 1993
Submitted by: Sam Ming Wang