CLUSTAL-X是一个图形化的多序列 比对工具 ,利用这个工具可以对数据 进行比对,除掉结构 相同的或者只有个别碱基序列不同的序列,最后对保留的结果得到最后对保留的结果得到“.phy”格式 文件 。PHYLIP软件 是一个免费的集成的进化分析 工具,有华盛顿大学 遗传 学系Joseph felsenstein 编写,1980年首次释放,目前已经升级到3.6.7版本。PHYLIP包含了35个程序 ,这些程序基本上囊括了系统 发生分析方面的所有方面,并且可以在很多平台上运行。包括分子 程序组、距离程序组、基因 频率组、连续字符组、不连续字符组和进化树绘制组。
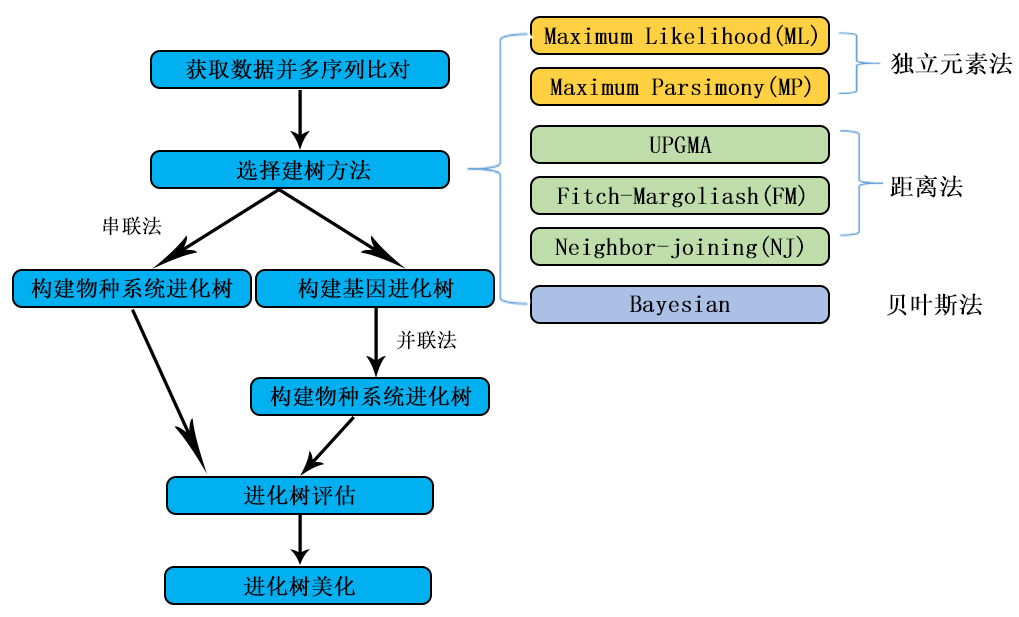
CLUSTAL-X 和PHYLIP软件相结合构建 分子进化树,具体的分析操作方法
(1) 用CLUSTALX软件对已知DNA 序列或者蛋白质 序列(如下)做多序列比对。
①点FILE进入Save sequence as,在format 框中选PHYLIP,文件在PHYLIP软件目录下以DNA.phy存在,点击OK,。
②将PHYLIP软件目录下的DNA.phy文件拷贝到EXE文件夹中, 用计事本方式打开的DNA.phy文件。
(2) 用PHYLIP软件推导进化树。
①进入EXE文件夹,点击SEQBOOT软件输入DNA.phy文件名,回车后,输入R更改参数,更改重复数字为200, 输Y确认参数。输入奇数种子3, 程序开始运行,并在EXE文件夹中产生outfile.
②把文件outfile改为infile, 点击DNADIST (PROTDIST for 蛋白 质序列)程序, 输入M更改参数,输入D选择data sets, 输入200。输Y确认参数, 程序开始运行,并在EXE文件夹中产生outfile。
③将outfile文件名改为infile,为避免与原先infile文件重复,将原先文件名改为infile1。
④在EXE文件夹中选择通过距离矩阵推测进化树的算法 ,点击NEIGHBOR程序(采用的是相邻连接(N-J)和UPGMAD相结合的算法), 输入M更改参数,输入D选择data sets, 输入200, 输入奇数种子3, 输Y确认参数, 程序开始运行,并在EXE文件夹中产生outfile和outtree两个结果输出。outtree文件是一个树文件,可以用treeview等软件打开。outfile是一个分析结果的输出报告,包括了树和其他一些分析报告,可以用记事本直接打开。
⑤将outtree文件名改为intree,点击DRAWTREE程序,输入font1文件名,作为参数。输Y确认参数。程序开始运行,并出现Tree Preview图。
⑥点击DRAWGRAM程序,输入font1文件名,作为参数。输Y确认参数。程序开始运行,并出现Tree Preview图。
⑦将EXE文件夹中的outfile文件名改为outfile1,以避免被新生成的outfile 文件覆盖。点击CONSENSE程序。输入Y确认设置。EXE文件夹中新生成outfile和outtree。Outfile文件用记事本打开。
⑧将EXE文件夹中的intree文件名改为intree1,将outtree改intree。点击DRAWTREE程序,输入font1文件名,作为参数。输Y确认参数。程序开始运行,并出现Tree Preview图。
⑨点击DRAWGRAM程序,输入font1文件名,作为参数。输Y确认参数。程序开始运行,并出现Tree Preview图。
2.2.3 TREEVIEW
Treeview 是一个从网免费下载 的读进化树免费软件,这个软件可以根据PHYLIP得到的树输出文件,做出无根树,有根树,还能在树中显示进化距离。
(责任编辑:admin)