简介
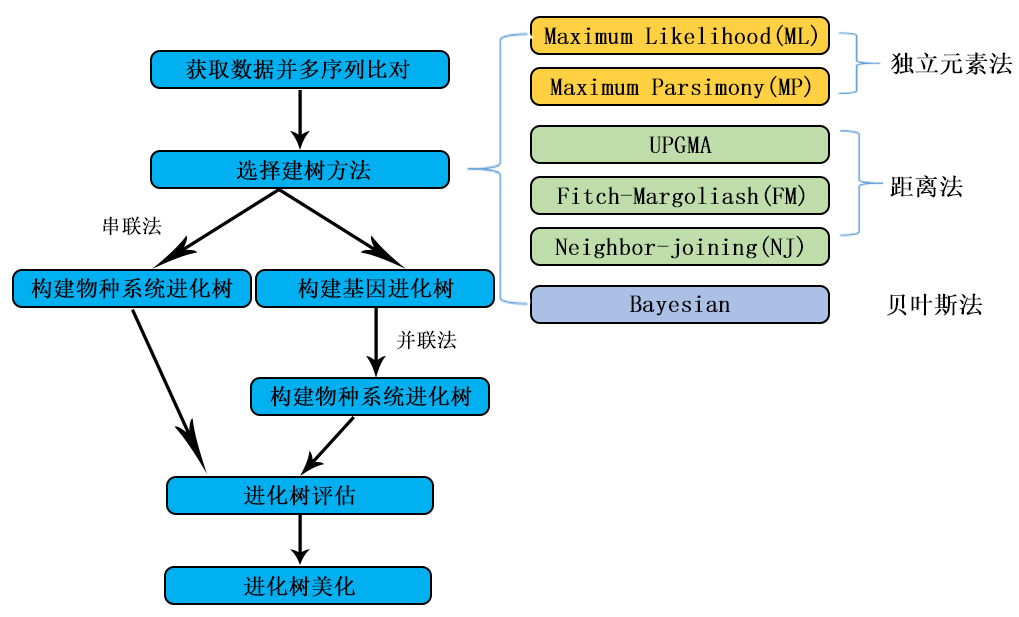
植物分子系统进化分析主要是利用携带植物遗传信息的生物大分子序列(比如 DNA 序列),釆用特定的数理统计算法来构建植物物种间的系统进化关系,并用一种树状分支图的形式来概括生物间的这种亲缘关系,即系统进化树。系统进化树可分为有根树和无根树,前者以外类群作为树根,后者无外类群树根。有根树的根节点为全部分类群的最近共同祖先,能够反映各分类群间的系统进化关系,而无根树仅反映出分类群间的分类关系。利用 DNA 分子序列进行植物系统进化分析是分子进化研究的必要手段。构建系统进化树的方法主要有距离法、最大简约法、最大似然法,以及贝叶斯推断法等。要解决特定植物类群间的系统进化问题,首先要挑选合理的分类群及 DNA 序列,尽量减少数据的偏倚,然后选择合适的构树方法,随后还要对结果进行评价,并给出生物学上的解释。
材料与仪器
步骤
植物分子系统进化树的构建的基本过程可分为如下几步:
(一)距离法
距离法是根据物种之间的进化距离进行系统进化树构建的一种方法。物种之间的进化距离一般取决于遗传模型;要根据不同 DNA 区域的进化模式选择最佳的核甘酸替换模型。一般若不考虑密码子的简并,并假定所有位点的替代速率相同,就能根据核昔酸替代模型估算出进化距离(如 JC 距离、Kimura 距离等)。
距离法中的邻位相连法(neighborjoining,NJ)是根据距离矩阵,在所有可能的拓扑结构中,选择分支长度和最小的作为最优树。通过对整个树的长度进行最小化,从而对树的拓扑结构进行限制。邻位相连法本质上是一种寻找最优拓扑结构的系统聚类算法,同时给出系统发育树的拓扑结构及分支的长度。其优点有:① 可以较快地构建系统树;② 适用于分析较大的数据集;③ 能够较方便地进行自展(bootstrap)检验。
(二)最大简约法
最大简约法(maximumparsimony,MP)最早是基于形态特征分类的需要发展而来,因算法不同而有许多版本。MP 法利用的只是对简约分析能提供信息的特征。一般在 DNA 序列数据分析中,利用的是有序列差异(至少有 2 种不同类型的核昔酸序列)的核昔酸位点,这些位点称为简约信息位点。利用 MP 法重建系统发生树,实际上是一个对给定序列其所有可能的树进行比较的过程。对某一个可能的树,首先对每个位点祖先序列的核昔酸组成作出推断,然后统计每个位点用来阐明差异的核昔酸最小替换数目。在整个树中,所有信息简约位点最小核昔酸替换数的总和称为树的长度。比较所有可能树,选择其中长度最小的树作为最终的系统树,即最大简约树。
(三) 最大似然法
最大似然法(maximum likelihood,ML)是评估所选定的进化模型能够产生实际观察到的数据的可能性。该方法明确地使用概率模型,其目标是寻找能够以较高概率产生观察数据的系统发生树。使用这种方法建树时,在每组序列比对中考虑了每个核昔酸替换的概率。例如,转换出现的概率大约是颠换的 3 倍。在 1 个 3 条序列的比对中,如果发现其中有 1 列为 1 个 C、1 个 T 和 1 个 G,则认为,C 和 T 所在的序列之间的关系更近。
设物种数为 N,对位排列后 DNA 或氨基酸序列的长度为 n,用这些序列组成的矩阵为
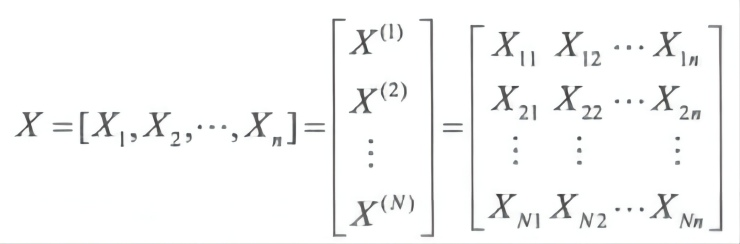
假定不同位点的进化是独立事件,根据该数据矩阵可以进行多种不同的似然估计。
1. 计算构树数据的似然率
对于一棵给定的树,可以用可能性得分评估所做出的假设,即评价所得到的系统发生树 n 对于给定的 1 组分类单元,假设它们的观察值为 M 为向量),可以选择一棵树,使得 F(M|T)最大,即最大似然法。
令 tvn 代表节点 v 和 u 以之间的分支长度,反映的是遗传距离或者进化时间。以概率 Px→y(tvn)表示在时间知内,从状态 x 转换到状态 y 的概率。假设有一个矩阵它是关于 n 个分类单元的实际观察值,M 描述每个分类单元四个特征的具体取值。同时假设存在一棵树 T,其叶节点(如 v、u)对应于这些分类单元,而树中的分支代表分类单元之间的距离 tvn,求该树的似然值 L = P(M|T)。
2. 计算树与子树的似然率
设长度为 n 的部分似然率矩阵为 q,定义 qi = Pix(t),这里,为树枝长度。有:

式中,Qi 为部分似然率的乘积。
3. 计算树枝长度的似然率
4. θ 的最大似然估计
θ 的最大似然为 maximize logL(θ|X,T)θ∈Θ,θ 满足
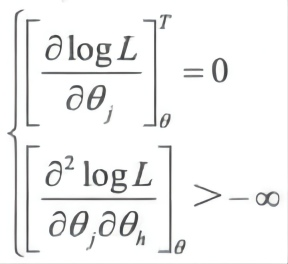
(四)贝叶斯法
贝叶斯法(Bayesian analysis)是釆用最大似然法的基本原理,同时引入马尔可夫链蒙特卡罗模型(Markov chain Monte Carlo analysis),使得构树时间和 ML 法相比大大缩短,能够胜任大数据集的分析。该方法最终得出的一棵基因树,是一组拥有相近最大似然率树的多数合意树,该树节点在这一组树中出现的百分率是该节点后验概率的近似值,故 Bayesian 法在构建一棵基因树的同时也给出了其节点的支持率。同以往的最大似然法相比,贝叶斯法的优越性在于:能够以很高的计算速度处理大型数据集,同时还提供了衡量树可信性的有效参数一后验概率。
(五)Mrbayes 软件构建系统发生树操作步骤
启动 Mrbayes 程序,依次输入命令:
(1)Execute filename. nex,打开待分析文件,文件必须和 Mrbayes 程序在同一目录下。
(2)Lset nst=6 rates=invgamma,该命令设置进化模型为 with gamma-distributed rate variation across sites 和 a proportion of invariable sites 的 GTR 模型。模型可根据需要更改。
(3)mcmc ngen = 1 000 000 samplefreq=1 000,保证在后面的可能性分布中(probability distribution)至少取到 10 000 个样品。默认取样频率:every 100th generationo 如果分裂频率(split frequencies)的标准偏差(standard deviation)在 1 000 000 代(generations)以后低于 0.01,当程序询问:"Continue the analysis?(yes/no)",回答 no;如果高于 0.01,yes 继续直到该值低于 0.01。
(4)sump bumin=2 500(在此为 10 000 个样品,即任何相当于取样的 25% 的值),参数总结(summarize the parameter),程序会输出一个关于样品(sample)的替代模型参数的总结表,包括 mean、mode 和 95% credibility interval of each parameter,要保证所有参数 PSRF(the potential scale reduction factor)的值接近 1.0,如果不接近,分析时间要延长。
(5)sumt bumin = 2 500,总结树(summarize tree)。程序会输出一个具有每一个分支的 posterior probabilities 的树,以及一个具有平均支长(mean branch lengths)的树。这些树会被保存在一个可以由 treeview 等读取的树文件中。
(六)系统发生树的可靠性检验
在系统发生推断中,常釆用一定的统计检验来分析获得的系统发生树的可靠性。一种是利用某一参量来对所获得树及其相近树进行结构差异检验。在 ML 法中常利用似然值,而在最小进化法中则利用所有支的总长度进行。这种方法是一种保守检验,而且检验的程序非常复杂,需要很大的计算机内存。另一种是分析每个内支可靠性,其中常用的方法有:① 标准误估计,即计算内支长度及其标准误,检验内支长度与 0 间的偏差,得到一个置信概率(confidence probability,CP),CP 值越高,支的长度也就越可靠。通常,当 CP ≥ 0.95 或 0.99 时,可认为该支的长度在统计上有效。② 自举检验(bootstrap test),这是一种重抽样技术,可用来估计在不知道或难以分析得到取样分布的情况下内支与统计有关的变异性。通过自举检验,可得到一个自举置信水平(bootstrap confidence level,BCL)。计算机模拟表明,当 BCL > 0.9 时,CP 值与 BCL 值二者非常相近,认为结果可靠。
来源:丁香实验