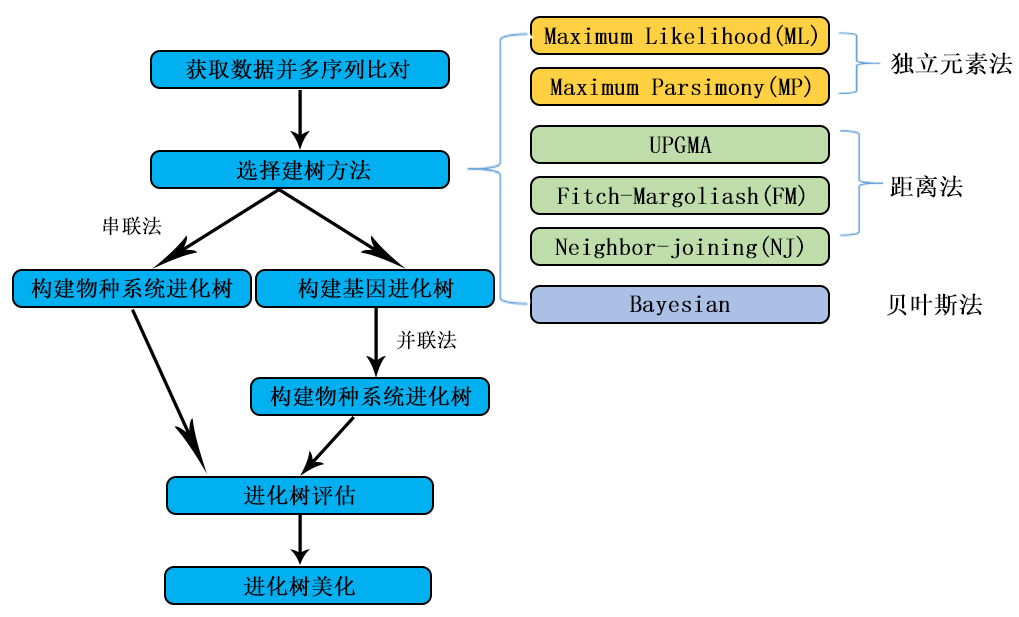
步一步教你如何做系统进化树
互联网
我在此介绍几个进化树分析及其相关软件的使用和应用范围。这几个软件分别是PHYLIP、PUZZLE、PAUP、TREEVIEW、CLUSTALX和PHYLO-WIN(LINUX)。 ⑴ 要对所分析的多序列目标进行排列(To align sequences)。做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW,前者是在WINDOW下的而后者是在DOS下的。 ⑵ 要构建一个进化树(To reconstrut phyligenetic tree)。构建进化树的算法主要分为两类:独立元素法(discrete character methods)和距离依靠法(distance methods)。所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。而距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离。独立元素法包括最大简约性法(Maximum Parsimony methods)和最大可能性法(Maximum Likelihood methods);距离依靠法包括除权配对法(UPGMAM)和邻位相连法(Neighbor-joining)。 ⑶ 对进化树进行评估。主要采用Bootstraping法。进化树的构建是一个统计学问题。我们所构建出来的进化树只是对真实的进化关系的评估或者模拟。如果我们采用了一个适当的方法,那么所构建的进化树就会接近真实的“进化树”。模拟的进化树需要一种数学方法来对其进行评估。不同的算法有不同的适用目标。 一般来说,最大简约性法适用于符合以下条件的多序列: i 所要比较的序列的碱基差别小, ii 对于序列上的每一个碱基有近似相等的变异率, iii 没有过多的颠换/转换的倾向,iv 所检验的序列的碱基数目较多(大于几千个碱基); 用最大可能性法分析序列则不需以上的诸多条件,但是此种方法计算极其耗时。如果分析的序列较多,有可能要花上几天的时间才能计算完毕。 UPGMAM(Unweighted pair group method with arithmetic mean)假设在进化过程中所有核苷酸/氨基酸都有相同的变异率,也就是存在着一个分子钟。这种算法得到的进化树相对来说不是很准确,现在已经很少使用。邻位相连法是一个经常被使用的算法,它构建的进化树相对准确,而且计算快捷。其缺点是序列上的所有位点都被同等对待,而且,所分析的序列的进化距离不能太大。 另外,需要特别指出的是对于一些特定多序列对象来说可能没有任何一个现存算法非常适合它。最好是我们来发展一个更好的算法来解决它。但无疑这是非常难的。我想如果有人能建立这样一个算法的话,那他(她)完全可以在Proc.Natl.Acad.Sci.USA.上发一篇高质量的文章。
i,DNA和蛋白质序列数据的分析软件。 ii,序列数据转变成距离数据后,对距离数据分析的软件。 iii,对基因频率和连续的元素分析的软件。 iv,把序列的每个碱基/氨基酸独立看待(碱基/氨基酸只有0和1的状态)时,对序列进行分析的软件。 v,按照DOLLO简约性算法对序列进行分析的软件。 vi,绘制和修改进化树的软件。在此,我主要对前两种功能软件进行说明。
图中的8和50分别表示8个序列和每个序列有50个碱基。然后,打开软件SEQBOOT: 图中的D、J、R、I、O、1、2代表可选择的选项,键入这些字母,程序的条件就会发生改变。D选项无须改变。J选项有三种条件可以选择,分别是Bootstrap、Jackknife和Permute。文章上面提到用Bootstraping法对进化树进行评估,所谓Bootstraping法就是从整个序列的碱基(氨基酸)中任意选取一半,剩下的一半序列随机补齐组成一个新的序列。这样,一个序列就可以变成了许多序列。一个多序列组也就可以变成许多个多序列组。 根据某种算法(最大简约性法、最大可能性法、除权配对法或邻位相连法)每个多序列组都可以生成一个进化树。将生成的许多进化树进行比较,按照多数规则(majority-rule)我们就会得到一个最“逼真”的进化树。Jackknife则是另外一种随机选取序列的方法。 它与Bootstrap法的区别是不将剩下的一半序列补齐,只生成一个缩短了一半的新序列。Permute是另外一种取样方法,其目的与Bootstrap和Jackknife法不同,这里不再介绍。R选项让使用者输入republicate的数目。所谓republicate就是用Bootstrap法生成的一个多序列组。根据多序列中所含的序列的数目的不同可以选取不同的republicate。当我们设置好条件后,键入Y按回车。得到一个文件outfile 这个文件包括了100个republicate。
选项O是让使用者设定一个序列作为outgroup。一般选择一个亲缘关系与所分析序列组很接近的序列作为outgroup(本例子不选outgroup),outgroup选择的好坏将直接影响到最后的进化树的好坏。选项M是输入刚才设置的republicate的数目。设置好条件后,键入Y按回车。生成两个文件outfile和treefile。
该文件包括了227个进化树。Treefile可以用TREEVIEW软件打开同样包含了这227个进化树。 键入Y按回车。生成两个文件outfile和treefile。Treefile用TREEVIEW打开。
我们看出两个树是同样的。但在outfile的树上的数字表示该枝条的Bootstrap支持率(除以100.6)。到现在,8个序列的进化树分析(最大简约法)已经完成。 选项D有四种距离模式可以选择,分别是Kimura 2-parameter、Jin/Nei、Maximum-likelihood和Jukes-Cantor。选项T一般键入一个15-30之间的数字。选项M键入100。运行后生成文件。 这个文件包含了与输入文件相同的100个republicate,只不过每个republicate是以两两序列的进化距离来表示。文件中的每个republicate都省略了第一排的Mo3 Mo5 Mo6 Mo7 Mo8 Mo9 Mo12 Mo13。以这个输出文件为输入文件,执行NEIGHBOR软件。 选项M键入100。生成两个文件outfile和treefile用记事本和TREEVIEW打开后,发现这两个文件都含有100个进化树。再将treefile文件更名后输入CONSENSE软件,又得到两个文件outfile和treefile,这就是最后的结果。以上是对DNA序列的分析,如果要对蛋白质序列进行分析,PROTDIST、PROTPARS等软件。其他软件的用法可以参照PHYLIP的documents。
|