定量蛋白质组学
互联网
2844
定量蛋白质组学
在分析正常组织或病理组织变化过程中(如在生长、分化、药物暴露或肿瘤发作期间)蛋白质的表达谱时,能够定量分析变化是非常重要的,明确地探测这些多肽经历的上调或下调过程也是很重要的。这些蛋白质有可能是药物作用的关键靶标,因此检测这些蛋白质对药物研发和医学处理至为重要。最早达到这个目标的方法是正确应用一些计算机程序(如Melanie或PDQuest)来分析比较这些组织(如正常组织和病理组织)的标准图。为了能够获得符合统计学要求的标准图,最少需要在每个状态下都收集3~5个样品;然后联合这些样品,在相同的实验条件下平行运行,同时产生至少5个二维图谱,这样做的目的是使误差最小化。精密复杂的计算机程序会合并这5张不同的图,并产生一个参考图和一个“病理”图,它们之间可能有相当程度的重叠。这样得到的系统比较图可以通过检测斑点上考马斯蓝的吸附量的变化而检测出上调或下调的蛋白质。尽管这个方法不但极为消耗人力而且过程也是漫长的,但是这个方法可靠。在最近这些年,其他的处理手段也被开发出来,主要是利用了有差别地对蛋白质的残基进行同位素标记的原理。涌现出的第一项技术称为同位素标记的亲和标签(isotope-coded affinitytag,ICAT),该技术是用化学试剂来修饰半胱氨酸残基的巯基基团(Gygi等,1999;Han等,2001)。这个化学试剂由三个区域组成:一个加到巯基基团上的终端碘尾巴;一个不是用d0 就是用d8 (d为氘,因而给予的两组不同标记的蛋白质在分子质量上有8Da的差别)标记的中间区域或连接区域;一个是同位素捕获的末端,这个末端包含了一个生物素钓饵。两个被标记的样品(不论是用轻试剂标记的还是用重试剂标记的)按1:1的比例混合,然后进行胰蛋白酶消化。这会产生一种非常不均一的混合肽,其全细胞溶出物的肽的数量可以达到几十万,这不适合直接进行质谱分析。为了减少这种巨大的样品复杂性,可以将混合物先上样至抗生物素蛋白柱上进行亲和纯化。只有含有半胱氨酸残基的肽(大约占全部肽的8%~9%)才能被捕获,进而进行质谱分析。每一个肽都被分成了相隔8Da的两个峰,分别代表了轻/重试剂的标记。这些双峰的比例代表的也就是它们的原始的样品混合物中的比例,从而可以提供这些多肽上调或下调的直接明细。但因为这些ICAT化学试剂的价格比较昂贵,所以最近Sechi(2002)和Gehanne等人(2002)推荐了一组价格比较便宜的试剂,也就是do/d:酰胺,这些试剂对于琉基基团施加了同样的效果,并且在进行ICAT时表现了相同的作用(具有这种附加的好处,即它们可以进行标准的二维图谱,也就是说,可以分析完整蛋白质,而不是蛋白质的消化肽)。尽管携带半胱氨酸残基进行同位素标记的两个肽的分子质量差别只有3Da,但这并不会给现代的质谱仪带来任何问题,因为现代质谱仪分析的质量准确度也能够为空间间隔这么小差别的邻近峰提供基准分辨率。
据Zhang和Regnier的报道(2002),在使用ICAT时也有恼人的缺点。Zhang和Regnier采用的捕获技术与ICAT技术的发明者报道的基于抗生物素蛋白―生物素亲和ICAT技术不同。对完全的细胞溶出物进行胰蛋白酶消化会得到复杂的混合物,在处理这种复杂的混合物时,应该想到使用不同的色谱方案,比如采用反相色谱后,再接着采用离子迁移分离。应该指出的是,如果通过一个反相柱来分离ICAT标记的肽,就会有一个让人头疼的同位素效应发生,也就是说氘化的肽比对应的没有氘化的肽洗脱得更早;这样的分离会导致同位素比例上的巨大变化,这种变化通过重构体的两个不同的洗脱谱可以看出。对于越小的标签肽,这种效应越明显。例如,在一个简单的、含有半胱氨酸的八肽的情况下,轻肽/重肽之间的色谱解析度可以高达R8 =0.74(需要说明的是,及,=1.2仅仅意味着基准的可分辨的峰)。因为柱洗出液是由ESI―MS连续分析的,所以通过同位素的比例来获得正确的定量的峰的比例成为一个特别困难的任务。Zhang和Regnier(2002)报道,在C18 柱中,有20%的消化肽,其同型重构体的分辨率超过了0.5。相反,他们又报道在用13 C和12 C琥珀酸酯差异标记肽的情况下,这种同位素效应会完全消失。所以他们强烈建议,试图在C18 柱分离时,使用这种类型的肽编码。Liu和Regnier(2002)建议使用整体编码策略,这个策略充分利用了蛋白质水解期间产生的氨基和羧基的差异衍生化。在胰蛋白酶消化期间产生的羧基基团将从H2 18 O中结合18 O。来自于对照样品和实验样品的伯胺,在与1 H3 ―N―乙酰氧基琥珀酰亚胺或2 H3 ―N―乙酰氧基琥珀酰亚胺进行蛋白酶水解之后,会被差异酰基化。
另外,其他的很多操作过程也有人报道。例如,Goodlett等人(2001)介绍了肽的甲酯化(用d0 甲醇或d3 甲醇),这个过程是将天冬氨酸和谷氨酸的侧链羧酸以及羧端的羧酸转化为甲酯。也有几个研究组(Hale等,2000;Keough,Lacey和Youngquist,2000;Beardsley,Karty和Reilly,2000;Brancia,Oliver和Gaskell,2000;Peters等,2001)建议将赖氨酸衍生为高精氨酸,以便于在利用MALDI-TOF-MS研究蛋白质/肽的时候提高强度。后来,Cagney和Emili(2002)采用了同样的衍生化程序,他们将这种处理途径命名为质量编码的丰度标签(mass-coded abundance tagging,MCAT),并通过差异胍基化来对胰蛋白酶消化的肽的羧端赖氨酸进行修饰(这会导致42Da的分子质量差别,而不是ICAT情况下的8Da)。
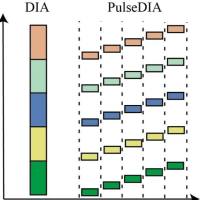
上述这些方法实际上是类似的,它们都利用了多肽之间的质量差别来推断多肽的定量表达比率。特殊的图像分析软件可以匹配图像、定量斑点、对信号归一化,并且通 过比较可提供任何一组两个蛋白质的表达差异。在图中可以看到一个DIGE分析的例子。
乳腺癌ErbB-2转化细胞经Cy3和Cy5染色(DIGE技术)的N―琥珀酰亚胺酯差异标记