【求助】4 kb片段连接pcDNA3.1+的问题
丁香园论坛
9033
各位高手,兄弟最近忙于质粒构建,意图将一段长约4000 bp的片段,通过BamH1和EcoR1两个酶切位点连接到pcDNA3.1+载体上。情况如下:
1. 通过LA Taq酶扩增,并纯化回收目的片段(引物中带有酶切位点,保护性碱基=3,上游带有Kozak序列);由于回收效率很高,所以在进行下一步双酶切时只加了12 μl的目的片段,H2O 20 μl,10 X K BUFFER 4 μl,两种酶各2 μl,37℃作用过夜(12-14 h),再进行胶回收。
2. 载体pcDNA3.1提取质粒后,也按照以上的体系进行双酶切,时间为6 h。
3. 将酶切后的木点片段和载体均取2 μl进行电泳,结果显示大小与预期相符,亮度相近。
4. 连接时,目的片段:载体=4--10:1(因亮度相近,所以按照体积比)都做过,10 μl体系和15 μl体系都尝试过。
5. 感受态为新制备的JM109,转化效率没有具体测过但别人用后均连接成功,阳性质粒眼观转化效率很高。
6. 限制性内切酶为TAKARA,T4连接酶是国内一家小公司的产品,不过实验室一直用,效果一直有保证,同期进行连接的其他同学也都连接成功。
结果:
转化后,平板上均有数量不等的菌落产生,少时4-5个,多时10来个。摇菌扩增8 h,进行菌液PCR检测(含阳性对照),检测组无阳性结果,但一直在1000 bp左右出现较弱条带,阳性组正常。提取质粒后,10 μl体系双酶切2 h,竟然在2000 bp和3000 bp处出现较弱条带(比Marker稍暗)。
特此悬赏10个叮当,请高手指点迷津。
如果有效果好的、适于长片段连接的方法,也请大家指教,多谢。
1. 通过LA Taq酶扩增,并纯化回收目的片段(引物中带有酶切位点,保护性碱基=3,上游带有Kozak序列);由于回收效率很高,所以在进行下一步双酶切时只加了12 μl的目的片段,H2O 20 μl,10 X K BUFFER 4 μl,两种酶各2 μl,37℃作用过夜(12-14 h),再进行胶回收。
2. 载体pcDNA3.1提取质粒后,也按照以上的体系进行双酶切,时间为6 h。
3. 将酶切后的木点片段和载体均取2 μl进行电泳,结果显示大小与预期相符,亮度相近。
4. 连接时,目的片段:载体=4--10:1(因亮度相近,所以按照体积比)都做过,10 μl体系和15 μl体系都尝试过。
5. 感受态为新制备的JM109,转化效率没有具体测过但别人用后均连接成功,阳性质粒眼观转化效率很高。
6. 限制性内切酶为TAKARA,T4连接酶是国内一家小公司的产品,不过实验室一直用,效果一直有保证,同期进行连接的其他同学也都连接成功。
结果:
转化后,平板上均有数量不等的菌落产生,少时4-5个,多时10来个。摇菌扩增8 h,进行菌液PCR检测(含阳性对照),检测组无阳性结果,但一直在1000 bp左右出现较弱条带,阳性组正常。提取质粒后,10 μl体系双酶切2 h,竟然在2000 bp和3000 bp处出现较弱条带(比Marker稍暗)。
特此悬赏10个叮当,请高手指点迷津。
如果有效果好的、适于长片段连接的方法,也请大家指教,多谢。
看了下你的情况,我有几个问题
1)为什么选用BamH1和EcoR1两个酶切位点?
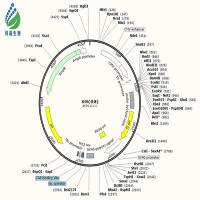
从pcDNA3.1+载体图谱上看,这两个酶切位点相隔太近,你用双酶切载体制备vector的时候,如何确保两个位点的酶切充分完全?我的建议是,如果不想换酶切位点,线性化载体的时候就采用单酶切的方法,先切EcoRI,取少量产物跑胶确定已经线性化,再切BamHI,胶回收酶切产物。另外,如果时间和经济允许,目的片段序列也允许,建议换用HindIII+XhoI这两个酶切位点,NEB的这两个酶是可以双酶切的,我用这两个位点已经构建了几十个constructs了,屡试不爽。
2)是否有设置自连对照?
这个问题实际上是上一个问题的延续,本质上就是检测载体双酶切的效率如何,根据自连对照克隆生长情况,还可以大致推断整个连接系统是否有效,以及感受态的效率如何。不知道你有否设置自连对照。如果有,那么自连对照的克隆生长情况如何?有没有提质粒跑胶看看片段大小?
3)我从来不用PCR来鉴定克隆。这个也跟个人喜好有关吧,我总觉得这种方法太容易产生cross contamination。我比较喜欢用in gel screening这个方法,省事省时,而且很直观。
4)最后友情提示一下,凡是构建constructs的所有电泳和胶回收操作,一定要使用新鲜的TAE buffer,而且一定要用新鲜的双蒸水配制。
4kb的insert虽然有点大,但是难度还不是最大。
祝你成功!
1)为什么选用BamH1和EcoR1两个酶切位点?
从pcDNA3.1+载体图谱上看,这两个酶切位点相隔太近,你用双酶切载体制备vector的时候,如何确保两个位点的酶切充分完全?我的建议是,如果不想换酶切位点,线性化载体的时候就采用单酶切的方法,先切EcoRI,取少量产物跑胶确定已经线性化,再切BamHI,胶回收酶切产物。另外,如果时间和经济允许,目的片段序列也允许,建议换用HindIII+XhoI这两个酶切位点,NEB的这两个酶是可以双酶切的,我用这两个位点已经构建了几十个constructs了,屡试不爽。
2)是否有设置自连对照?
这个问题实际上是上一个问题的延续,本质上就是检测载体双酶切的效率如何,根据自连对照克隆生长情况,还可以大致推断整个连接系统是否有效,以及感受态的效率如何。不知道你有否设置自连对照。如果有,那么自连对照的克隆生长情况如何?有没有提质粒跑胶看看片段大小?
3)我从来不用PCR来鉴定克隆。这个也跟个人喜好有关吧,我总觉得这种方法太容易产生cross contamination。我比较喜欢用in gel screening这个方法,省事省时,而且很直观。
4)最后友情提示一下,凡是构建constructs的所有电泳和胶回收操作,一定要使用新鲜的TAE buffer,而且一定要用新鲜的双蒸水配制。
4kb的insert虽然有点大,但是难度还不是最大。
祝你成功!
要先确定你那么长的片段当中不含有这两个酶的酶切位点。但一直在1000 bp左右出现较弱条带,应该是非特异条带。还有就是酶切产物最好都进行电泳后回收,以免载体切下的片段又连回载体上,降低了成功率。如果实在不行的话,考虑TA克隆。
BamHI、EcoRI可以的,我做过。
载体和片段都是胶回收的?在回收片段纯度没有问题下考虑以下因素1、 克隆长得少可能表示载体酶切尚可,很可能片段酶切得不好。合成的引物是否完整?上面的酶切位点是否正确?如果时间允许,克隆到T载体上再酶切。很多PCR产物酶切连接不上的问题克隆到载体上后再酶切都没有问题了。
2、片段测过序吗?内部是否有这两个酶?
载体和片段都是胶回收的?在回收片段纯度没有问题下考虑以下因素1、 克隆长得少可能表示载体酶切尚可,很可能片段酶切得不好。合成的引物是否完整?上面的酶切位点是否正确?如果时间允许,克隆到T载体上再酶切。很多PCR产物酶切连接不上的问题克隆到载体上后再酶切都没有问题了。
2、片段测过序吗?内部是否有这两个酶?
tozitong1983,感谢您的回答,很专业。
1. bamh1和ecor1这两个酶是我个人比较偏爱的酶。我构建过十多个重组质粒,几乎都是用这两种酶,一直也没出现过问题(pet30a载体上这两个酶是紧挨着的,重组质粒构建的也很顺利。)我查了载体谱图,hind3我用不了,而且bamh1和ecor1离的不算近,差了10多个bp。实验室一直用takara的酶,没有neb那么好,呵呵。
2. 自连对照已经补充了,结果还没有出。
1. bamh1和ecor1这两个酶是我个人比较偏爱的酶。我构建过十多个重组质粒,几乎都是用这两种酶,一直也没出现过问题(pet30a载体上这两个酶是紧挨着的,重组质粒构建的也很顺利。)我查了载体谱图,hind3我用不了,而且bamh1和ecor1离的不算近,差了10多个bp。实验室一直用takara的酶,没有neb那么好,呵呵。
2. 自连对照已经补充了,结果还没有出。
wangch84 wrote:
tochenjy1974和dorianmell站友,目的片段中不含有这两种酶切位点序列。
已经开始进行ta克隆
转化的结果都不稳定,有时候长的多有时候长得少,之前曾经就这个问题发帖向大家请教过,有战友反应这一步其实很简单,没什么很难得因素。但是不管怎样,我现在对长片段的TA克隆是有心理阴影了,所以我一般都是直接设计带酶切位点的PCR引物,扩增出目的片段后直接酶切再连目的载体,至今还没有做不出来的。
再次祝你好运,有进展记得与大家分享。
4KB的PCR产物片段直接双酶切克隆,要靠运气了。强烈建议TA克隆,测序OK后,再换载体,这样的话成功率才会高。
楼主需要确认几点:
1,PCR产物两端的酶切位点在不在;
2,筛选阳性克隆时,不管是菌落PCR还是质粒酶切,由于片段与太大,都不好去判断。建议:如果是PCR鉴定,则可以在中间设计一条引物,与目的载体的一条通用引物进行配对,扩增出来的大小为1k左右,这样便于判断。如果采取酶切方法鉴定,建议在片段中选取内切酶,只要切出来的大小符合我们理论上的要求即可;
3,克隆时阴性对照非常重要。
希望对你有帮助
楼主需要确认几点:
1,PCR产物两端的酶切位点在不在;
2,筛选阳性克隆时,不管是菌落PCR还是质粒酶切,由于片段与太大,都不好去判断。建议:如果是PCR鉴定,则可以在中间设计一条引物,与目的载体的一条通用引物进行配对,扩增出来的大小为1k左右,这样便于判断。如果采取酶切方法鉴定,建议在片段中选取内切酶,只要切出来的大小符合我们理论上的要求即可;
3,克隆时阴性对照非常重要。
希望对你有帮助
todolphin_705_200再次感谢你的回答。
载体对照组的转化结果,只长了一个菌落,应该还可以吧
载体对照组的转化结果,只长了一个菌落,应该还可以吧
tozitong1983,KAPA HIFI酶一定很贵吧,呵呵
载体对照组的转化结果,只长了一个菌落
载体对照组的转化结果,只长了一个菌落
wangch84 wrote:
tozitong1983,KAPA HIFI酶一定很贵吧,呵呵
载体对照组的转化结果,只长了一个菌落
自连对照组只有一个菌落,我觉得不是太好,可能你的感受态效率不是太高。一般双酶切的载体,自连对照应该有十来个甚至几十个菌落出现,不过要比加了insert的那一碟少得多。所以我觉得你有必要再检查下你的感受态是否有问题。
当然,连接体系的效率偏低也可能出现这样的情况。
[来自丁香园WAP]
最新进展,终于将4kb片段连到宝生物t载体了,也双酶切回收了目的片段进行连接。转化长了8个菌落,扩增后碱法小提质粒,双切结果却是在2000多和1000那有两条带,很奇怪啊
最新进展,终于将4kb片段连到宝生物t载体了,也双酶切回收了目的片段进行连接。转化长了8个菌落,扩增后碱法小提质粒,双切结果却是在2000多和1000那有两条带,很奇怪啊
tozitong1983最新进展,终于将4kb片段连到宝生物t载体了,也双酶切回收了目的片段进行连接。转化长了8个菌落,扩增后碱法小提质粒,双切结果却是在2000多和1000那有两条带,很奇怪啊
wangch84 wrote:
tozitong1983最新进展,终于将4kb片段连到宝生物t载体了,也双酶切回收了目的片段进行连接。转化长了8个菌落,扩增后碱法小提质粒,双切结果却是在2000多和1000那有两条带,很奇怪啊
如果这一步没问题,那么恭喜你,终于迈出成功的一步。
你的第二步,看起来有些问题,8个克隆效率明显偏低。小提质粒后双酶切size也不对,即使自连的情况下size也不对。建议,
1)检查你的酶是否有问题?可以找一个已知的含有这两个酶切位点的质粒过夜酶切再跑胶看看size是否正确,酶切是否完整。
2)请再次确认你的感受态效率ok,没有污染。
3)重新酶切制备insert和vector,然后连接转化。整个过程注意我上面提到的那些细节。
如果还是做不出来,一个简单的建议,换实验室别的人来做吧,或许换个人试试很快就搞定。你也正好可以比较下自己的问题出在哪里
祝你好运。
不成熟的看法:如果一切都正常,包括载体,可能是PCR产物有问题。PCR鉴定用的引物是目的片段扩增用的引物吗?
to coffeeshine,是的
tozitong1983我先用的片段是从T/A克隆的质粒上切下来的,载体是从另外一个阳性质粒上切下来的
T载体上的正确吗?最好是测序验证一下。
我初步判断你从T载体上切下来的片段就有问题,所以你再与其它载体连接后长出的克隆酶切就有问题。大片段的酶切很容易混淆。
TAKARA的T载体大小有3K左右,你4K的片段和3K的片段比较难分开。如果T载体质粒测序OK的话,你需要将片段切下来这一步做准确了。建议你用合适浓度的琼脂糖胶分离,约1.5%左右,跑胶时间可能在2小时以上。
我初步判断你从T载体上切下来的片段就有问题,所以你再与其它载体连接后长出的克隆酶切就有问题。大片段的酶切很容易混淆。
TAKARA的T载体大小有3K左右,你4K的片段和3K的片段比较难分开。如果T载体质粒测序OK的话,你需要将片段切下来这一步做准确了。建议你用合适浓度的琼脂糖胶分离,约1.5%左右,跑胶时间可能在2小时以上。
你连接了多久啊
咪咪蘑萝 wrote:
你连接了多久啊
wangch84 wrote:
你好,这个重组质粒构建了将近一个月,最后成功了。期间一直用TOP10高效率感受态,但是检测结果一直出现莫名其妙的问题,考虑可能是质粒与宿主菌之间有一些奇妙的关系。后来换成高效率的JM109感受态,一次就连接成功。
咪咪蘑萝 wrote:
我也要做分子克隆方面的,不懂的地方还希望得到你的指教
wangch84 wrote:
好的,大家互相交流
我上次将连接产物拿过去测序了,结果发现有一个碱基发生了突变。会是什么原因呢?
咪咪蘑萝 wrote:
我上次将连接产物拿过去测序了,结果发现有一个碱基发生了突变。会是什么原因呢?
在PCR过程中,发生碱基突变是常事。任何一种高保真酶都不可能完全保证不发生突变;越好的酶,发生突变的几率越低。
仅仅发生一个突变还好,如果突变之后,氨基酸没变,就继续用吧。
如果你想矫正一下,可以看一下一下突变的位置。如果就在目的片段的5`端或3`端,可以通过设计引物的方式矫正;如果在序列内部,可以通过SOE PCR或者买一些定点突变的试剂盒。
同学你好,这个帖子都好久了,不过我菜鸟一个刚开始做,也是连接4000+的片段,我想连到pcDNA3.1上,两个多月也没成功,到T载体也连不上去,不知道您有什么意见!
idoctorwhoi wrote:
同学你好,这个帖子都好久了,不过我菜鸟一个刚开始做,也是连接4000+的片段,我想连到pcDNA3.1上,两个多月也没成功,到T载体也连不上去,不知道您有什么意见!
1. 感受态种类与效率,尤其转化效率,一定要很高。效率越高转化成攻率越高。有的时候片段与载体已经连接成功了,只不过在转化时,自身空间结构较大,没有转进去。
2. 酶切反应:如果可以,请用thermo fisher的内切酶,无论单酶切还是双酶切,6 h足够了。takara的酶批次间稳定性经常有问题,而且长时间酶切,片段很容易切碎。
3. 4000+的片段先加A连接到T载体上,利用片段两端的酶切位点双切,回收后再连接载体。
4. 如果第3步还是不行,请尝试一下这个偏方:找一个新载体p,长度要比pcDNA3.1还要长,先练到载体p上,切下来再连接pcDNA3.1。。。。。。。这个方法,不是常规protocol,我当初遇到一些问题时,出此下策,竟然成功。
预祝实验顺利!
本文由丁香园论坛提供,想了解更多有用的、有意思的前沿资讯以及酷炫的实验方法的你,都可以成为师兄的好伙伴
师兄微信号:shixiongcoming