蛋白免疫印迹(Western Blot)技术手册
互联网
36027
1.简介
Western Blot中文一般称为蛋白质印迹。它是分子生物学、生物化学和免疫遗传学中常用的一种实验方法。其基本原理是通过特异性抗体对凝胶电泳处理过的细胞或生物组织样品进行着色。通过分析着色的位置和着色深度获得特蛋白质在所分析的细胞或组织中的表达情况的信息。
2.原理
Western Blot与Southern印迹杂交或Northern印迹杂交方法类似,但Western Blot采用的是聚丙烯酰胺凝胶电泳 ,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。经过PAGE分离的蛋白质样品,转移到固相载体(例如硝酸纤维素膜NC膜 )上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技术也广泛应用于检测蛋白水平的表达。其图解为:

Western Blot显色的方法主要有以下几种: i. 放射自显影 ii. 底物化学发光ECL iii. 底物荧光ECF iv. 底物DAB呈色。现常用的有底物化学发光ECL和底物DAB呈色,体同水平和实验条件的是用第一种方法,目前发表文章通常是用底物化学发光ECL。只要买现成的试剂盒就行,操作也比较简单,原理如下(二抗用HRP标记):反应底物为过氧化物+鲁米诺 ,如遇到HRP,即发光,可使胶片曝光,就可洗出条带。
3.简略步骤

4. 蛋白样本提取制备
蛋白样品制备是Western Blotting的第一步,更是决定WB成败的关键步骤,总体原则和注意事项:
(1)尽可能提取完全或降低样本复杂度只集中于提取目的蛋白(通过采用不同提取方法或选择不同的试剂盒产品)
(2)保持蛋白的处于溶解状态(通过裂解液的PH 盐浓度 表面活性剂、还原剂等的选择)
(3)提取过程防止蛋白降解、聚集、沉淀、修饰等,(低温操作,加入合适的蛋白酶和磷酸酶抑制剂)
(4)尽量去除核酸,多糖,脂类等干扰分子(通过加入核酸酶或采取不同提取策略)
(5)样品分装,长期于-80 ℃中保存,避免反复冻融。
4.1细胞裂解
我们公司有针对细胞内的不同蛋白专门的蛋白裂解液试剂盒供您选择。
4.11细胞裂解操作方法:
(1)培养的细胞经预冷的PBS漂洗2次,裂解液中加入蛋白酶和磷酸酶抑制剂。
(2)吸净PBS,加入预冷的裂解液,((1 ml per 107 cells/100mm dish/150cm2 flask; 0.5ml per 5x106 cells/60mm dish/75cm2 flask).
(3)用细胞刮子刮取贴壁细胞,将细胞及裂解液温和地转移至预冷的微量离心管中,
(4)4℃摇动30 min。
(5)4℃离心12,000 rpm,10 min(根椐细胞种类不同调整离心力)
(6)轻轻吸取上清,转移至新预冷的微量离心管中置于冰上,即为蛋白样本,弃沉淀.
4.2组织裂解
(1)用灭菌的预冷的工具分离目的组织,尽量置于冰上以防蛋白酶水解,
(2)将组织块放在圆底的微量离心管或Eppendorf管中,加入液氮冻结组织于冰上均质研磨,长期可保存于-80 °C。
(3)每约5 mg加入约300 μl 预冷的裂解液lysis buffer,冰浴匀浆后置于4℃摇动2小时,裂解液体积与组织样本量有适当比例,(最终的蛋白浓度至少达到0.1 mg/ml, 理想的蛋白浓度应为1-5 mg/ml).
(4)4℃离心12,000 rpm,20 min,
轻轻吸取上清,转移至新预冷的微量离心管中置于冰上,即为蛋白样本,弃沉淀。
4.3蛋白酶和磷酸酶抑制剂
推荐购商品化蛋白酶和磷酸酶抑制剂复合试剂盒或COOKTAIL,或按下表配制:
备注:其中Sodium orthovanadate配制活化方法如下:
所有步聚均需在通风橱中进行:
(1) 用双蒸水配制100 mM 正矾酸钠溶液.
(2)用盐酸HCl 调至pH 9.0
(3)煮沸至溶液无色,尽量减少水分挥发.
(4)冷却至室温
(5)再调pH 至 9.0
(6)再煮沸至无色
(7)重复上述过程,直至溶液煮沸冷却后达pH 9.0
(8)用水定容至原体积
(9)分装保存于- 20 °C. 溶液变黄则弃之不用.
5. 蛋白定量
我们公司有不同蛋白定量的试剂盒,可满足不同实验员的需求。Bradford 法 Lowry 法或BCA 法 (操作简单、需分光光度计或酶标仪),小牛血清白蛋白 (BSA) 作为标准曲线。如果裂解液中有NP40或其它表面活性剂,则推荐使用BCA 法。三种方法或产品比较
列表如下:

6. 电泳上样样品的准备
6.1变性、还原蛋白样本
一般的抗体只能识别抗原蛋白中的部份序列结构(表位),因此,为使抗体能够达到结合该表位而需要将蛋白样本进行变性,使之打开折叠的空间结构,
蛋白变性一般使用含阳离子变性去污剂如SDS的上样buffer(loading buffer),并于95-100 °C 煮沸 5分钟,对于多次跨膜蛋白,可以于70 °C 加热 5-10分钟,SDS的阴离子环绕蛋白肽键使之带负电荷,蛋白分子量不同,结合的SDS数量不同,所带负电荷也不同,电泳迁移速度不同,因此SDS-PAGE电泳可将不同分子量的蛋白分离开。
6.2天然和非还原样本
某些抗体识别的表位是非连续氨基酸构成的蛋白三维结构,此种情况则需要进行非变性的WB,抗体的说明书一般会标注,这种非变性电泳不加SDS,样本也不需煮沸。
某些抗体仅识别蛋白的非还原态,如某些cysteine基的氧化态,因此loading buffer和电泳液中不加入β-mercaptoethanol and DTT。

7. 电泳
7.1 PAGE胶的制备
不同分子量的蛋白选择不同的凝胶浓度(参考下表),原则上高分子量蛋白用低浓度胶,低分子量蛋白用高浓度胶分离。

不同浓度的凝胶的配制方见公司提够的相关试剂盒说明书,首先配制各组分贮备液,然后分别配制浓缩胶和分离胶。
7.2蛋白分子量Marker
预染或非预染各种分子量的蛋白,用于标示电泳中蛋白的大小和示踪
7.3阳性对照
目的蛋白或明确表达目的蛋白的组织或细胞的蛋白提取物,用于检验整个实验体系和过程的正确性有效性/特别是一抗的质量和效率。建议使用该对照。可查阅文献或抗体说明书选择购买或自提该对照样本。
7.4内参对照
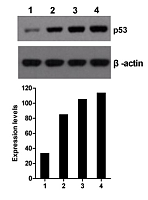
管家基因编码的、很多组织和细胞中都稳定表达的蛋白,用于检测整个WB实验过程及体系是否正常工作,并作为半定量检测目的蛋白表达量的标准对照。必须设立。

7.5上样与电泳
每孔上样量为20-40 μg蛋白,使用专用枪头或注射针头,勿溢出加样孔,电泳时间按电流仪说明书推荐方法使用,(1小时或过夜,取决于电压大小),当染料到达胶的底部,关电源停止电泳,胶不能存放,应立刻进行下一步的转膜。
8. 转膜与显色(Western Blot)
8.1胶中蛋白的检测
电泳后检测蛋白是否迁移正确与平均,可采用铜染或考马斯蓝染色检测,如果凝胶中的蛋白需要进行转膜则需可逆的铜染法,否则采用不可逆考马斯蓝法染色。
铜染法:电泳胶用蒸馏水洗数秒钟,加入0.3 M CuCl2染色5-10分钟,再用去离子水洗一次,在暗背景下观察在兰色胶背景下蛋白出现透明条带,胶置于0.1- 0.25 M Tris/0.25 M EDTA pH 8.0缓冲液中漂洗脱色两次,再置于电转缓冲液中开始转膜。
考马斯蓝法:用40%双蒸水, 10% 醋酸, 50%甲醇的溶液固定胶中蛋白,考马斯蓝R-250染液(凯基产品)室温染色4小时至过夜,保持摇匀,转入67.5%双蒸水, 7.5% 醋酸, 25%甲醇l摇匀至脱去多余的染料,蛋白被染成深蓝色。
8.2蛋白转膜
蛋白因结合SDS而带电荷,在电场下从胶中转至膜上,转膜操作根椐电转仪制造商的说明书进行转膜方式分为半干和湿转两种,半干式转膜速度快,而湿式成功率高并特别适合用于分子量大于100KD的蛋白。
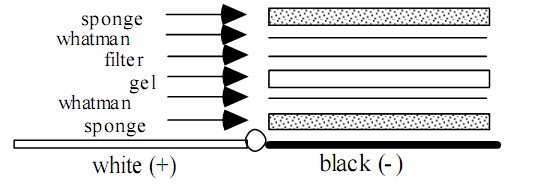
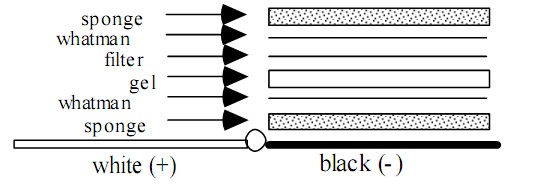
湿式转膜三明治排列为:海绵/纸/胶/膜/纸/海绵,全部紧密排列,特别是胶/膜之间不能留有气泡,三明治安放的方向确认正确负极方为带负电的胶里的蛋白,向正极方(膜)电迁移。标准的电转缓冲液为1X Tris-glycine buffer 不含SDS,但加入20%甲醇,如果转膜的蛋白分子量大于80KD,则推荐加入SDS使之终浓度为0.1%。
半干式转膜中,三明治的排列为:/纸/胶/膜/纸,用电转缓冲液浸湿后,直接置于电转仪的正负极之间。胶于负极而膜置于正极。半干式的电转缓冲液可不同于湿式的电转缓冲液,推荐为:48 mM Tris, 39 mM glycine, 0.04% SDS, 20%甲醇,
两类膜可供选择:硝酸纤维素膜和PVDF膜(正电荷尼龙膜),根据不同需要选择(下表),PVDF膜需要浸泡甲醇中1-2分钟,再孵育于冰冷的电转缓冲液中5分钟,胶也需在冰冷的电转缓冲液中平衡3-5分钟,否则转膜时会导致条带变形。

注:大蛋白和小蛋白的转膜
电转移缓冲液中SDS与甲醇的平衡、蛋白的大小、胶的浓度都会影响转膜效果,如下调整可以增加转膜效率:
大蛋白(大于100 KD)
1.对于大蛋白而言,其在凝胶电泳分离迁移较慢,而从凝胶转出也非常慢,因此对于这种大分子量蛋白应该用低浓度的凝胶,8%或更低,但因低浓度的胶非常易碎,所以操作时需十分小心,
2.大蛋白易在凝胶里形成聚集沉淀,因此;转膜时在电转移缓冲液加入终浓度为0.1%的SDS,以避免出现这种情况,甲醇易便SDS从蛋白上脱失,因此应降低电转移缓冲液中甲醇的浓度至10%或更低,以防止蛋白沉淀。
3.降低电转移缓冲液中甲醇的比例以促进凝胶的膨胀,易于大蛋白的转出。
4.如果使用PVDF,甲醇是必需的,且转膜前PVDF需用甲醇活化。但如果是NC膜,甲醇可以不必加入电转移缓冲液中。
5.选择湿式,4 ℃转膜过夜,以取代半干式转膜。
小蛋白(小于100 KD)
1. SDS妨碍蛋白与膜的结合,特别是对小蛋白更是如此,因此,对于小分子的蛋白,电转移缓冲液中可以不加SDS。
2.保持20%的甲醇浓度
对于大于500KD的蛋白,请参考下述文献:Bolt and Mahoney, High-efficiency blotting of proteins of diverse sizes following sodium dodecyl sulfate–polyacrylamide, gel electrophoresis. Analytical Biochemistry 247,185–192 (1997).
更多的转膜注意事项:
Ø避免用直接接触膜,应使用镊子,手指上的油脂与蛋白会封闭转膜效率并易产生背景污斑
Ø排列三明治时,尽量用移液器或15ml试管赶除胶与膜之间的气泡,或将三明治放在装有的培养皿中以防止气泡产生,请戴手套!
Ø确认裁剪的膜和滤纸与凝胶尺寸相同,否刚导致电流不能通过膜,从而转膜无效
Ø鸡抗体易于与PVDF膜和其它尼龙膜结合,导致高背景,请替换成硝酸纤维素膜以降低背景。
Ø膜上蛋白的检测:丽春红。为检测转膜是否成功,可用丽春红染色,2%的丽春红贮备液(20ML):: 2%丽春红(0.4克)溶于30%三氯乙酸(6克)和30%磺基水杨酸(6克)丽春红染色工作液:2%的丽春红贮备液1:10稀释,即加9 倍的ddH2O染色方法:将膜放入TBST洗一次,再置于丽春红染色工作液中,在室温下摇动染色5分钟,大量的水洗膜直至水变清无色蛋白条带清晰,(膜也可以用TBST或水重新洗后再进行染色)PVDF膜需用甲醇再活化后用TBST洗后进行封闭。
9. 膜的封闭
为防止一抗或/和二抗与膜的非特异性结合产生的高背景,因此需要进行膜的封闭,
传统上有两种封闭液:脱脂奶粉或BSA,脱脂奶粉成本低但不能用于磷酸化蛋白(因脱脂奶粉含有酪蛋白,该蛋白本身就是一种磷酸化蛋白),使用脱脂奶粉会结合磷酸化抗体从而易产生高背景。
某些抗体用BSA封闭时因不明原因可能会产生比脱脂奶粉更强的信号,,请仔细阅读说明书注明的注意事项和膜的特殊的封闭方法。
配制5%脱脂奶粉或BSA 溶液:每100 ml TBST中加入5 g脱脂奶粉或BSA,混匀后过滤,如不过滤会导致使膜污染上细微黑颗粒。
封闭时,4°C摇动,封闭1 hour ,再用TBST洗5秒,进入下一步抗体的孵育。
10. 一抗的孵育
孵育Buffer:按抗体说明书建议的稀释倍数,用TBST稀释一抗,如果说明书没有建议的稀释倍数,则参照一般推荐的稀释倍数(1:100-1:3000),一抗浓度过高会导致产生非特异性条带。
某些实验室传统上在封闭液中孵育抗体,而有些实验室用不含封闭剂的TBST来孵育抗体,结果因抗体而异,有时两者结果相同,有时结果不同。
某些实验室传统上在封闭液中孵育抗体,而有些实验室用不含封闭剂的TBST来孵育抗体,结果因抗体而异,有时两者结果相同,有时结果不同。
注:如果不存在高背景的问题,某些抗体用含低浓度(0.5 – 0.25%)脱脂奶粉或 BSA的封闭液来,稀释,可产生相对更强的信号条带。
孵育时间:一抗的孵育时间可从几小时至过夜(一般不超过18小时)不等,取决于抗体与蛋白的亲和性和蛋白的含量丰度,建议使用较高的抗体稀释倍数和较长的孵育时间来保证特异性结合。
孵育温度:尽可能低温孵育,如果在封闭液中孵育一抗过夜,应在4oC进行否则会产生污染而破坏蛋白(降别是磷酸基团)。孵育一抗时需保持适当的摇动使之均匀覆没膜,防止结合不均匀。
11. 二抗的孵育
一抗孵育结束后,用TBST摇动洗膜数次,每次5min或更长,去除残留的一抗。
孵育Buffer和稀释倍数:用TBST按说明书推荐的倍数稀释二抗,如果说明书没有标出稀释倍数,则按常规的倍数稀释(1:1000- 1:20,000)预试,二抗的浓度过高也会导致非特异性条带。亦可以在封闭液中孵育二抗(和一抗),但可能在降低背景同时导致特异性条带的信号也减弱,可能是封闭剂阻碍了抗体与靶蛋白的结合。
孵育时间和温度:室温、1-2 hours, 摇动。
二抗连接物:推荐使用二抗连接HRP,不建议连接AP碱性磷酸酶,因其不够灵敏。
12. 显色
显色分为酶促底物发光和化学发光法或荧光法
酶促底物发光法代表为DAB显色法,与其它同类方法的比较如下图所示:

而现在最常用的是化学发光法:HRP化学发光底物 Luminol(ECL法chemiluminescence)及其改良法,
13. 常见问题分析与解决方案



13.2其他常见问题分析
无信号
一抗和二抗不匹配
二抗需和一抗宿主的物种相同(如一抗来自兔,二抗为抗兔抗体)。
没有足够的一抗或二抗结合目标蛋白
使用高浓度抗体。4°C延长孵育时间(如过夜)。
封闭剂与一抗或二抗有交叉反应
使用温和的去污剂如吐温-20,或更换封闭剂(常用的脱脂奶粉、BSA、血清或明胶)。
一抗不识别待检物种的蛋白
参照说明书,比对免疫原序列和蛋白序列以确保抗体和目的蛋白会发生反应,设置阳性对照。
抗原量不足
每条泳道蛋白上样量不低于20-30?μg,使用蛋白酶抑制剂并设置阳性对照。
组织中目的蛋白含量低
浓缩使信号最大化(例如检测核蛋白可用核裂解物)。
转膜不充分
使用可逆染色剂如丽春红S?检测转膜效果,检查转膜操作是否正确。PVDF?膜需预先浸在甲醇中,然后浸到转移缓冲液中。
洗膜过度
勿过度洗膜。
过度封闭使目标蛋白不能显色
使用0.5%奶粉或无奶粉代替5%奶粉的抗体稀释液,或更换封闭剂,减少封闭时间。
一抗失效
使用新鲜抗体,重复使用有效浓度会降低。
二抗受叠氮钠抑制
避免叠氮钠和HRP?标记抗体一起使用。
检测试剂盒过期和底物失活
使用新鲜的底物。
背景高
未进行非特异性封闭或封闭不充分
延长封闭时间,考虑更换合适的封闭剂。Abcam推荐5%脱脂奶粉、3% BSA或血清封闭30?分钟,这些可以包含在抗体缓冲液中。
一抗浓度过高
稀释抗体至合适浓度,以更高稀释度抗体延长孵育时间(耗时长但特异性结合最好)。
抗体孵育温度过高
4°C孵育。
二抗与封闭剂非特异性结合或反应
设置二抗对照(不加一抗)。
二抗与封闭剂非特异性结合或与封闭剂反应
在孵育和洗涤液中加入温和去污剂如吐温-20。脱脂奶粉含有酪蛋白,该蛋白本身就是一种磷酸化蛋白,会结合磷酸化特异性抗体而易产
生高背景。使用BSA代替奶粉作为封闭剂。
未结合蛋白质洗涤不充分
增加洗涤次数。
选膜不当产生的高背景
硝酸纤维素膜比PVDF膜背景低。
膜干燥
在孵育过程中防止膜变干,每一步都要保证膜有充分的反应液,可放入搅拌子不断搅动或轻轻振荡使膜浸在溶液里。
多带现象
细胞传代次数过多,蛋白表达不同
使用原始未传代的细胞株,和现在的细胞株一起做平行对照实验。
体内表达的蛋白样本具有多种修饰形式如乙酰化、甲基化、烷基化、磷酸化、糖基化等
参考文献,使用试剂使样品去磷酸化、去糖基化来证明翻译后的修饰。
蛋白样本降解(蛋白质分子量降低)
在样品缓冲液中加入足够的蛋白酶抑制剂。
检测到未经报道过的新蛋白或同一蛋白家族中具有相似表位而结构不同的蛋白
查阅其它文献报道,或BLAST搜寻,使用说明书推荐的细胞株或组织。
一抗浓度过高,高浓度时常出现多条蛋白带
降低抗体浓度和/或孵育时间。
二抗浓度过高,高浓度产生非特异性结合
降低抗体浓度,加入二抗对照(不加一抗)。
抗体未经纯化
使用亲和纯化的抗体,减少非特异条带。
条带为非特异性条带
应用封闭多肽来区分特异性和非特异性条带,只有特异性条带能被封闭从而消失。
目标蛋白形成多聚体
SDS-PAGE电泳加样前,煮沸10分钟而不是5分钟,使蛋白质解聚。
背景有不均匀的白色斑点
转膜时膜上有气泡或抗体在膜上分布不均
转膜过程中尽量除去气泡,抗体孵育时保持摇动。
背景有黑色斑点
抗体结合了封闭剂
过滤封闭剂。
深背景出现白色条带
一抗或二抗加入过多
稀释抗体的浓度。
分子量标准条带呈黑色
抗体和分子量标准发生了反应
在分子量标准和第一个样品之间增加一个空白条带。
目的条带染色过低/过高
分离不彻底
改变凝胶比例:分子量大的蛋白用低浓度胶,分子量小的蛋白用高浓度胶。
条带“微笑”效应
1.迁移过快
2.电泳温度过高(改变了pH值和迁移速度)
2.电泳温度过高(改变了pH值和迁移速度)
降低迁移速度或低温电泳(冷库或冰上)。
相同的蛋白杂交出现大小不均匀条带
制备凝胶时凝胶凝固太快,致使泳道中丙烯酰胺的比例不均匀
参照凝胶的配方,在凝胶中加入适量TEMED,放置时在凝胶顶部加入适量0.1% SDS(水稀释)以防凝胶变干。
凝胶染色不均匀
1.细菌污染
2.抗体量不足
(1)4 °C保存抗体并使用新鲜的缓冲液浸泡凝胶。
(2)确保振荡条件下孵育膜或抗体充分覆盖膜。