支原体污染的特点及检验
互联网
最近看大家常提到支原体污染,就翻了翻书,做了些笔记,现在贴出来和大家分享:
支原体是一种大小介于细菌和病毒之间(最小直径0.2um)并独立生活的微生物。约有1%可通过滤菌器。支原体无细胞壁形态呈高度多形性。可为圆形、丝状或梨形。
支原体形态多变,在光境下不易看清内部结构。电镜下观察支原体膜为三层结构。其中央有电干密度大的密集颗粒群或丝状的中心束。支原体多吸附或散在于细胞表面和细胞之间。横断面与细胞微绒毛相似,但微绒毛电子密度比支原体小,且中央无颗粒群或中央束。
支原体代谢需固醇类物质、部分种类需要精氨酸、O 2或葡萄糖,每一种支原体都有自身持点。多数支原体适合于偏碱条件下生存(PH7.6—8.0),对酸耐受性差。对热比较敏感,对一般抗生素不敏感。
支原体污染细胞后。培养液可不发生混浊。多数情况下细胞病理变化轻微或不显著,细微变化也可由于传代、换液而缓解。因此易被忽视。但个别严重者,可致细胞增殖缓慢。甚至从培养器皿脱落。
为确定有无支原体污染可做如下检酗:
①相差显微境检测:将细胞按种于事先放置于培养瓶内的盖玻片上,24h后取出,用相差油镜观察。支原体呈暗色微小颗粒位于细胞表面和细胞之间。
②荧光染色法:荧光染色用能与DNA特异性结合的荧光染料Hoechst 33258,可使支原体内含有的DNA着色,染色后用荧光显微镜观察。具体方法如下:首先将细胞接种盖玻片上,在细胞未长满前取出玻片,用不合酚红的Hanks液漂洗一下,用l: 3醋酸甲酵固定10Min,然后用生理盐水漂洗。置于用生理盐水配浓度为50pg/ml的Hoechst 33258中染色10min。染色后用蒸溜水洗1—2min。向细胞面滴加pH5.5磷酸缓冲液数滴,然后置荧光显微镜下观察。镜下支原体为散在于细胞同围或附于细胞膜表面的亮绿色小点。
③电镜检查;用扫描电镜方法简便快速。也可以利用透射电镜。
④DNA分子条文检查或支原体培养等方法:检出率高。但方法较为复杂
支原体是一类缺乏细胞壁的原核细胞型微生物,大小一般在0.3-0.5um之间,呈高度多形性,有球形、杆形、丝状、分枝状等多种形态。它不同于细胞,也不同于病毒。支原体用普通染色法不易着色,用姬姆萨染色很浅,革兰染色为阴性。支原体可在鸡胚绒毛尿囊膜上或细胞培养中生长,营养要求比细菌高。
细胞培养(特别是传代细胞)被支原体污染是个世界性问题。在细胞培养中支原体感染发生率达到63%,支原体感染发生后能改变细胞的DNA,RNA及蛋白表达,又不能通过可视法对其进行检测,而且它对细胞的生长率影响较小,不易引起注意。国内外研究表明,95%以上是以下四种支原体:口腔支原体(M.orale)、精氨酸支原体(M.arginini)、猪鼻支原体(M.hyorhinis)和莱氏无胆甾原体(A.laidlawii),为牛源性。以上是最常见的污染细胞培养的支原体菌群,但能够污染细胞的支原体种类是很多的,国外调查证明,大约有二十多种支原体能污染细胞,有的细胞株可以同时污染两种以上的支原体。
支原体污染的来源包括工作环境的污染、操作者本身的污染(一些支原体在人体是正常菌群)、培养基的污染、污染支原体的细胞造成的交叉污染、实验器材带来的污染和用来制备细胞的原始组织或器官的污染。
组织细胞培养工作中,主要从以下几个方面来预防支原体的污染:控制环境污染;严格实验操作;细胞培养基和器材要保证无菌;在细胞培养基中加入适量的抗生素。支原体污染细胞后,特别是重要的细胞株,有必要清除支原体,常用方法有抗生素处理、抗血清处理、抗生素加抗血清和补体联合处理。支原体最突出的结构特征是没有细胞壁,一般来讲,对作用于细胞壁生物合成的抗生素,如 -内酰胺类、万古霉素等完全不敏感;对多粘菌素(polymycin)、利福平、磺胺药物普遍耐药。对支原体最有抑制活性及常用于支原体感染治疗的抗生素是四环素类、大环内酯类及一些氟喹诺酮;其他类抗生素如氨基糖苷类、氯霉素对支原体有较小抑制作用,所以常不用来作为支原体感染的化学治疗剂。InvivoGen公司研究开发的新一代支原体抗生素M-Plasmocin能有效地杀灭支原体,不影响细胞本身的代谢,并且用M-Plasmocin处理过的培养细胞,不会重新感染支原体。
红霉素有效,而且不影响细胞生长。网友“smilewf”经验是细胞刚开始被支原体污染,细胞生长极为缓慢,而且状态不好,细胞之间及底部总有一些亮晶晶的颗粒状物质,换液后好些,但过2天又会增多,培养液清亮,严重时象一层细沙铺在瓶底。刚开始以为是消化过头引起的细胞的碎片和细胞内的颗粒,后来发现与胰酶消化无关。使用红霉素(就在医院的门诊购买,很便宜)后这种现象明显好转,背景干净,细胞生长很快,状态明显好转,但好像红霉素不能完全根治支原体感染,停药后一段时间可以复发,但如果细胞很重要有没有多余的细胞时可以考虑一试,我用后觉得效果不错。
网友“zllxash”的经验:支原体是细胞培养中最常见的,干扰实验结果的一种污染。但由于不易被察觉,有些污染的细胞仍在继续使用。据查,目前各实验室使用的二倍体细胞和传代细胞中约有11%的细胞受到支原体污染。因此,对支原体污染应严加防范。
用卡那霉素预防支原体污染有效。
检测方法:
1、相差显微镜检测;
2、低张处理地衣红染色观察;
3、DNA荧光染色法;
4、电镜检测;
5、3-H胸腺嘧啶掺入法;
6、聚合酶链反映法;
7、免疫学方法;
8 、支原体的培养。
对于支原体污染的细胞,可以采取补救措施:
1、抗生素排除法:可以用5-10倍于常用量的冲击法,加入高浓度抗生素后作用24-48小时,再换入常规培养液,有时可能有效。推荐用量:庆大霉素 200ug/ml ,四环素10ug/ml ,卡那霉素50ug/ml 。对于支原体污染抗生素应用,预防性应用比污染后使用好。
2、 加温除菌:根据支原体耐热性能差的特点。将受支原体污染的细胞置于41度中作用5-10小时可以杀灭支原体。但由于41度对培养细胞本身也有较大的影响,故处理前,应先进行预试验,确定出最大限度杀伤支原体而对细胞影响较小的处理时间。
支原体菌株来源:M.Arginini ATCC23838 精氨酸支原体,M.FermentaneATCC19989发酵支原体 ,M.SalivariumATCC23064唾液支原体 ,M.HominisATCC23114人型支原体 ,M.OraleATCC23714口腔支原体 ,M.HyorhinisATCC29052猪鼻支原体。其共同引物序列来自16s和23s保守区域,外部引物为F1 5′-ACACCATGGGAGCTGGTAAT-3′,R1 5′-GTTCATCGACTTTCAGACCCAAGGCAT-3′;内部引物为 F2 5′-GTTCTTTGAAAACTGAAT-3′,R2 5′-GCATCCACCAAAAACTCT-3′。
PCR反应体系和条件:10×缓冲液(10mMTris-HCl、500mMKCl、20mMMgCl2、0.01%明胶)、PrimerF1、R1、F2、R2的浓度为2nmol/μL、2.5mMd NTPs、Taq酶、H2O、石蜡油覆盖。反应温度和时间为:94℃/2min预变性、94℃/30s变性、55℃/1min退火、72℃/1min延伸,循环30次后72℃延伸5min。第1次PCR反应取模板10μL,第2次PCR反应以第1次PCR扩增产物为模板取1μL。
一种名为BM-Cyclin的化学试剂,除体外培养癌细胞中支原体污染具体方法:
1、选用抗支原体药物(BM-Cyclin-1、2 ),磷酸缓冲液配制,溶液抽滤除菌,0~4℃保存。
2、细胞处理过程:细胞传代同时加BM-Cyclin-1 10μg/ml,37℃培养3天;胰蛋白酶消化、传代,加入BM-Cyclin-2 5μg/ml,37℃培养3天;继续传代追加BM-Cyclin-2 5μg/ml一次,培养2~4天(如细胞污染严重可重复上述处理过程)弃药液,D-Hank’s液洗2次,胰蛋白酶消化传代,进行常规培养。
3 、在细胞检测或实验前,最好再用常用量的5倍庆大霉素或卡那霉素作短时间处理,每天30~60分钟。
支原体的污染是目前细胞实验室比较普遍存在的问题,用药物来杀灭支原体虽是一种重要措施,毕竟是一种补救方法,而且清除后,支原体仍可重新污染细胞。
因此,重要的还在于预防,如严格选用无支原体污染的牛血清,保持细胞培养间洁净和严格无菌操作等。
网友“arlbor”把从网上和书上看到的一些资料与大家讨论一下吧:
细胞株若受到细菌、真菌、支原体、或是特定病毒等之污染时,会严重的影响实验的结果。而细菌、真菌等之微生物污染时,较易自培养基等的外观变化察觉。但是若受到支原体之污染时,细胞之外观较无明显变化,但是其污染会造成细胞之生长速率缓慢、细胞产生病变之型态改变等等变化。
各国细胞库支原体污染的统计表,如下:
国家 受支原体污染(%) 报告年度
USA-FDA 15 1993(past 30 years)
USA-ATCC 15-20 1992
Japan-(IFO,RIKEN,JCR 80~30~22 1981 1987-19931998
Germany-DSM 36 1990-1994
Argentina 65 1987
Israel 32 1986-1993
China 95 1990
造成支原体高污染率的原因为:
1、支原体size 0.1~0.8 um,无细胞壁,可透过一般过滤膜(0.22-0.45 um)
2、支原体污染时,没有明显的肉眼或一般光学显微镜可观察到的特征变化
3、过去缺乏简单、快速且可靠的检测方式
4、细胞流通间缺乏品管,造成实验室间的相互污染
5、研究或操作人员忽略污染问题
而支原体污染了来源:
1、已受污染的细胞
2、操作人员的疏失
3、已受污染的培养基、血清
4、操作环境不良、实验器具不洁等
由于支原体为人体口腔中之正常菌丛,实验室之操作人员的污染亦可能为污染源,须十分注意。而万一细胞已受支原体污染时,最佳的处理原则为将细胞高压灭菌后丢弃,以免污染其它洁净的细胞株。但是若是坚持要作去除支原体污染,有以下几种方法,只不过结果会与原始的细胞特性有差异。
去除支原体污染:
1、抗生素处理:BM-cyclin(Roche),MRA(mycoplasma removal agent,ICN),Ciprofloxcin(Bayer),Enrofloxacin(Bayer)
2、Nucleic acid metabolites:5-bromouracil,Hoechst 33258,bromodeoxyuridine
3、Anti-sera
预防与控制方面可从以下各点加强注意:
G 设备方面:
1、使用已作支原体测试ok之细胞株
2、于另一隔离之区域操作未知是否有支原体污染之细胞
3、使用不添加抗生素的培养基培养细胞
G 检测方面:定期以标准的支原体检测方法检测细胞、培养基、血清、ddH2O有否被支原体污染。
G 实验操作人员之无菌观念与无菌操作技术之要求
有关支原体污染之问题与回答:
o 应如何避免细胞污染?
细胞污染的种类可分成细菌、酵母菌、霉菌、病毒和支原体。 主要的污染原因为无菌操作技术不当、操作室环境不佳、污染之血清和污染之细胞等。 严格之无菌操作技术、清洁的环境、与品质良好之细胞来源和培养基配制是减低污染之最好方法。
o 如果细胞发生微生物污染时, 应如何处理?
直接灭菌后丢弃之
o 支原体 (mycoplasma) 污染的细胞, 是否能以肉眼观察出异状?
不能。 除极有经验之专家外, 大多数遭受支原体污染的细胞株, 无法以其外观分辨之。
o 支原体污染会对细胞培养有何影响?
支原体污染几乎可影响所有细胞之生长参数,代谢及研究之任一数据。故进行实验前,必须确认细胞为 mycoplasma-free,实验结果之数据方有意义。
o 侦测出细胞株有支原体污染时, 该如何处理?
直接灭菌后丢弃,以避免污染其它细胞株。
支原体之检测:
· 缺点:培养时间长,须3-5星期才能判断。有些支原体不能在培养基培养出来 (例如M. hyorhinis)。需同时培养支原体株作为正反应对照组,可能会造成污染。
· 材枓:dextrose、L-arginine HCl、Difco PPLO broth (Difco 0554-17-1) horse serum、15% yeast extract solution (autoclaved)、Bacto agar、无菌有盖螺旋试管(autoclaved)、无菌培养皿﹙60x15mm﹚、L-玻棒、37℃培养箱、厌氧缸(厌氧缸长期培养易有污染,所以厌氧包应用无菌水,每次打开厌氧缸后,用70% ethanol擦拭内壁。)
· 培养基制备:基本配方为Difco PPLO broth(60%), 马血清(20%), 15% yeast extreat solution (10%), 10x stock solution (10%)。由于培养时间长,可加入penicillin(50u/ml)至10x stock solution或加入thallium acetate (1:2000)至培养基中以防止其它细菌污染。
· 10x stock solution (1 liter) :称取50 g dextrose,10 g L-arginine HCl,溶于1 liter蒸馏水中。以0.22μm无菌过滤膜过滤灭菌。分装100 ml至瓶中,保存于 -70℃。
· 液体培养基 (1 liter) :称取21 g之Difco PPLO broth,0.02 g phenol red 于600ml蒸馏水中,加热搅拌溶解之。灭菌121℃,15分钟。待温度降低至室温后,于无菌操作台内加入200ml马血清,100ml之15% yeast extract solution,及100ml解涷之10x stock solution,混合均匀后,分装至已灭菌之有盖螺旋试管中,10ml/管。保存于4℃,期限1个月。
· 固体培养基 (1 liter ):称取21 g之Difco PPLO broth,0.02 g phenol red,15g Bacto agar于600ml蒸馏水中,加热溶解之。灭菌121℃,15分钟。放在50℃水中浴中,待温度降低至50℃时,于无菌操作台内加入200ml马血清,100ml之15% yeast extract solution 及100ml解冻之10x stock solution,混合均匀后,倒入60x50㎜之无菌培养皿中,5ml/培养皿。保存于4℃,期限1个月。
步骤:
· 取1 ml细胞培养液或0.2ml细胞悬浮液,接种于液体培养基中,置于37℃培养二周,观察是否有混浊或是pH值改变之情形。培养一周及二周后,分别取0.1 ml液体培养液涂在agar plate上,将其倒放置于37℃厌氧箱培养,培养至少3星期,持续观察是否有支原体之菌落出现。
· 另取1 ml细胞培养液或0.2ml细胞悬浮液,涂抹在agar plate上,37℃厌氧培养3星期,持续观察是否支原体菌落出现。
· 另作正负反应对照组,正反应对照组为Acholeplasma laidlawii (ATCC 23206) 与M. arginini (ATCC 23838)。负反应对照组为待测细胞之新鲜培养基。
结果判读:
· 支原体生长较慢,所以先在液体培养基培养1~2周增殖后,再培养于agar plate上。需至少培养3星期后,才可断定是否有支原体污染。所以整个测试需5个星期才知正确结果。
· 典型之支原体菌落类似荷包蛋(fried- egg like),乃是由于支原体往agar下层生长所致,为圆形无色透明菌落,大小约10-55μm。但是并非所有的支原体皆为似荷包蛋菌落,有些是圆形菌落或是类似粘菌类形态(slime)。
· 若要区分细胞或是气泡造成的类似菌落,可将其自agar plate上切下,重新培养于液体培养基中,培养一星期后再种入agar plate,若是细胞则不会生长。
DNA萤光染色法
· 原理︰利用萤光染剂(bisbenzimide, Hoechst 33258)侦测支原体污染。此染剂会结合到DNA之Adenosine-Thymidine (A-T) rich区域,因为支原体之DNA中A-T含量占多数(55~80%),所以可将其染色而侦测。被支原体污染之细胞经染色后,在细胞核外与细胞周围可看到许多大小均一之萤光小点,即为支原体之DNA,证明有支原体之污染。
· 测试步骤用间接步骤,将待测细胞悬浮液或细胞培养液接种于指示细胞培养液中(indicator cell,例如Vero cell or 3T6 cell),然后培养指示细胞再作萤光染色。正负反应对照组亦可以接种于indicator cell内作为对照。
· 待测细胞亦可作直接测试,但需为吸附型细胞,培养后直接作萤光染色。有些细胞易有萤光背景,而干扰结果判读,则建议使用接种于指示细胞之步骤。
· 特点︰简单、经济与灵敏,广泛使用,可作为例行之侦测步骤。可以侦测不易培养之支原体,例如M. hyorhinis,较直接培养法快,约一星期即可知道结果。
· 缺点:有时仍会有萤光背景,影响判读。
材料︰
· Hanks’ balanced salt solution without NaHCO3 or phenol red (HBSS, GibcoBRL 21250-014﹚、bisbenzimide (Hoechst No.33258, Sigma B-2883)、thimerosol (Sigma T-5125)、0.1M citric acid、02M disodium phosphate、glycerol、glacial acetic acid、methanol、strerile H2O、chamber slide (Nunc 177380)或chamber slide替代物:35 or 60mm sterile plate内放置无菌22x22mm盖玻片﹙浸于酒精内,使用前过火灭菌﹚,一般吸附型细胞均能吸附在盖玻片上。染色后取出盖玻片,细胞面朝下放在玻片上观察。
· Indicator cells: Vero (African green monkey kidney, ATCC CCL-81 or CCRC 60013) or 3T6(mouse fibroblast, ATCC CCL-96 or CCRC 60070),细胞须先测试无污染。αMEM with 10%FBS ( medium for Vero cell)
配制试剂:
· 无菌HBSS︰配制Hanks’ balanced salt solution,以0.22mm无菌过滤膜过滤灭菌。勿用高压蒸汽灭菌。
· Hoechst 33258 stock solution(100X)︰称取5.0 mg bisbenzimide和10.0 mg thimersol溶于100 ml无菌HBSS,置于棕色瓶中,室温下以电磁搅拌器搅拌30分钟至完全溶解后,以1ml分装至1.5ml小离心管中,贮存于-20℃。
· Hoechst 33258 working reagent︰取1 ml Hoechst 33258 stock solution,加入sterile 100 ml HBSS中,室温下搅拌45分钟,置于棕色瓶中并以铝箔纸包覆,避免光照,贮存于4℃。
· mounting solution︰22.2 ml 0.1 M citric acid和27.8 ml 0.2 M Na2HPO4加入于50 ml glycerol中混合,以1 N NaOH调整pH至5.5(pH很重要),贮存于4℃。
· 固定溶液(fixative)︰冰醋酸与甲醇以1︰3之体积比例混合,使用前才配制。
步骤:
· 培养Vero细胞: Vero细胞可作长期培养或制作多个冷冻保存管,测试前解冻培养。测试前一周培养Vero细胞。
· 在接种测试样品前一日,以trypsin-EDTA溶液处理Vero细胞,制成细胞悬浮液,以1~2x10^4 cells/ml培养于chamber slide中,每个well加入1ml细胞悬浮液,于37℃, 5%CO2培养。隔日确定生长良好后,即可接种测试样品。
· 接种测试样品:样品种类:1ml待测之培养基,1ml待测细胞培养液(可将细胞稍微刮下﹚,0.05~0.1ml冷冻管之细胞悬浮液。
· 将测试样品加入chamber slide内,培养5天后,进行Hoechst 33258的萤光染色观察。
· 以Acholeplasma laidlawii ATCC 23206 感染Vero细胞作为正反应对照组﹙或是已感染支原体之细胞培养液或冷冻细胞液﹚,负反应对照组为Vero cell之新鲜培养基( αMEM +10%FBS)。
DNA萤光染色检测︰
· 取出培养5日之Vero细胞,吸除培养液。于每个well中加入2 ml 1:3混合的冰醋酸/甲醇固定液,静置10 min后,吸除固定液。重复上述之步骤,风干10分钟。
· 于每个well中,加入1 ml Hoechst 33258 working solution,室温下静置30 min。
· 吸掉Hoechst 33258染液后,以无菌水洗涤3次,风干后加入1滴的mounting solution,并以盖玻片覆盖之。
· 以100x-400x萤光显微镜观察。打开水银光源10 min后,以UV激发光束(330~380 nm)之滤光镜,观察细胞核外是否有蓝色萤光小点或是丝状点之萤光物产生。
结果判读︰
若有支原体污染,在Vero细胞核外与细胞表面会出现蓝色萤光小点或是丝状点。其形状一致,与细胞残片染成之不规则点状物不同。其萤光可维持数星期。
图例 (A)为negative result : 萤光显微镜下,只观察到Vero细胞的细胞核,测试样品没有支原体的污染。(为positive result : 在Vero细胞核外与细胞表面会出现蓝色萤光小点和丝状点,表示测试样品有支原体的污染。

Positive Result 在Vero细胞核外与细胞表面会出现蓝色萤光小点和丝状点,表示测试样品有支原体的污染。

 污染测试--支原体 :
污染测试--支原体 :
PCR方法原理:
利用具特殊专一性之primers,经由PCR反应来复制mycoplasma DNA。所用之primers来自mycoplasma之conserved 16S-23S rRNA序列,由于此段spacer之序列依mycoplsma种类不同而不同,因此可依所复制之DNA大小及其restriction fragment大小差异来作侦测与鉴定。
特点:灵敏(e.g. 0.1~1.6 CFU / 5 ul sample) 与快速(一天)。可侦测不易培养之mycoplasma (e.g. M. hyorhinis)。不需培养mycoplasma作为正反应对照组,避免可能之污染。
缺点︰PCR反应很灵敏,易有伪阳性结果。此方法尚在评估中,故结果仅作为参考和内部品管之一部份。
材料与设备:
ATCC mycoplasma detection kit:可作50~100 reactions.
提供1st stage primer mixture与2nd stage primer mixture
positive control DNA (A. laidlawii ; M. pirum)
PCR reagents :
Taq polymerase ( 5 U / ul )
dNTP( dGTP, dCTP, dTTP, dATP, 1.25 mM each )
10x PCR buffer (with 1.5mM MgCl2)
25mM MgCl2
sterile ddH2O
Machine / Equipment:
PCR thermal cycler
agarose电泳设备
DNA电泳胶体观察设备
无菌PCR反应管
无菌1.5 ml微量离心管
无菌2ul, 20ul, 200ul, 1000ul Tips/ Pipetmans
方法:
此PCR反应是一种nested PCR,包括2阶段PCR反应,1st stage PCR结束后,取其反应物作2nd stage PCR,然后取2nd PCR产物作agarose gel electrophoresis,依DNA band之有无及片段大小来分析结果。
结果:使用UV light观察,并照相记录。

方法:
此PCR反应是一种nested PCR,包括2阶段PCR反应,1st stage PCR结束后,取其反应物作2nd stage PCR,然后取2nd PCR产物作agarose gel electrophoresis,依DNA band之有无及片段大小来分析结果。
结果:使用UV light观察,并照相记录。
方法再详尽点:
3.1 1st stage PCR reaction(total volume 25 ul):
3.1.1 测试样品 ( 2 ul / each )
3.1.1.1 直接取样测试细胞之培养液。
3.1.1.2 Positive control : mycoplasma DNA ( A. laidlawii ; M. pirum )
3.1.1.3 Negative control : ddH2O
3.1.2 Reaction mixture ( 23 ul / each )
3.1.2.1 10x PCR buffer (含1.5 mM MgCl2 ) 2.5 ul
3.1.2.2 1st stage primer mixture 0.5 ul
3.1.2.3 dNTP ( 1.25 mM each ) 1.0 l
3.1.2.4 MgCl2 ( 25 mM ) 0.5 ul
3.1.2.5 Taq DNA polymerase ( 5 U/ul ) 0.1 ul
3.1.2.6 ddH2O 18.4 ul
3.1.3 PCR program ( 1st PCR与2 nd PCR相同):使用PCR thermal cycler
3.1.3.1 step 1 : denaturation: 94 ℃ 30 sec
3.1.3.2 step 2 : denaturation: 94 ℃ 30 sec
3.1.3.3 step 3 : annealing: 55 ℃ 2 min
3.1.3.4 step 4 : extension: 72 ℃ 2 min
3.1.3.5 重复3.1.3.2至3.1.3.4步骤30 cycles
3.1.3.6 step 5 : final extension 72 ℃ 5 min
3.2 2 nd stage PCR reaction(total volume 25 ul)
3.2.1 测试样品:1st stage PCR product(1 ul / each)。
3.2.2 Reaction mixture ( 24 ul / each )
3.3.2.1 10x PCR buffer (含1.5 mM MgCl2 ) 2.5 ul
3.3.2.2 2 nd stage primer mixture 0.5 ul
3.3.2.3 dNTP ( 1.25 mM each ) 1.0 ul
3.3.2.4 MgCl2 ( 25 mM ) 0.5 ul
3.3.2.5 Taq DNA polymerase ( 5U/ul ) 0.1 ul
3.3.2.6 ddH2O 19.4 ul
3.2.3 PCR program同3.1.3;使用PCR thermal cycler
4.0 胶片电泳分析
4.1 胶片 (2.0%) 置备: 将2 g agarose溶于100 ml ( 1x ) TAE buffer。
4.2 电泳液:(1x) TAE buffer(Tris-acetate/EDTA electropheresis buffer)
4.3 选择100~500 bp DNA size Marker:取100 bp ladder Marker 5 ul ( 25 ng/ul )
4.4 取10 ul 2 nd stage PCR产物分析,各加入2 ul ( 6x ) loading dye。
4.5 进行电泳分离100V, 25 min。
4.6 胶片染色:ethidium bromide染色10 min,H2O退染10mim。(EtBr为致癌物质,请戴手套并小心操作)
4.7 结果:使用UV light观察,并照相记录。
Figure 1 : Agarose gel electrophoresis of the 2nd -stage PCR Products from eight commonly encountered Mycoplasma and A. laidlawii Species. ( from ATCC mycoplasma detection kit)
Table 1 : Variations of restriction fragment lengths of the 16S-23S rRNA intergenic spacer regions of commonly encountered species of mycoplasma. (from ATCC mycoplasma detection kit)
_No_restriction_site~Kbr_~H~M *_When_a_polyacrylamide_gel_is_used_for_the_resolution_of_amplified_DNA_products~E_a_double~Fband_product_with_one_bp_difference_may_be_observed_for_M._hominis._This_feature_can_serve_as_a_good_indicator_for_differentiated_M._hominis_from_M._arginini~E_which_only_produces_a_single~Fband_DNA_product.~K~Hp~M~2~1~Kp~M 
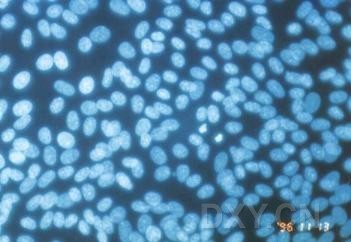
网友“zllxash”提供支原体污染的细胞在光学显微镜下的照片:如下








Protocol:Detection of Mycoplasma by Culture
1. Inoculate 2 agar plates with 0.1ml of test sample.
2. Inoculate 1 agar plate with 100cfu M. pneumoniae.
3. Inoculate 1 agar plate with 100cfu M. orale.
4. Leave 1 agar plate un-inoculated as a negative control.
5. Inoculate 1 broth with 0.2 ml of test sample.
6. Inoculate 1 broth with 100cfu M. pneumoniae.
7. Inoculate 1 broth with 100cfu M. orale.
8. Leave 1 agar plate un-inoculated as a negative control.
9. Incubate agar plates anaerobically for 14 days at 37°C using a gas jar with anaerobic gas
pak and catalyst.
10. Incubate broths aerobically for 14 days at 37°C.
11. Between 3 and 7 days and 10 and 14 days incubation, subculture 0.1 ml of test broth onto
an agar plate and incubate plate anaerobically as above.
12. Observe agar plates after 14 days incubation at x300 magnification using an inverted
microscope for the presence of mycoplasma colonies (see Figure 14).
Criteria for a Valid Result
All positive control agar plates and broths show evidence of mycoplasma by typical colony
formation on agar plates and usually a colour change in broths.
All negative control agar plates and broths show no evidence of mycoplasma.
Criteria for a Positive Result
Test agar plates infected with mycoplasma show typical colony formation.
Criteria for a Negative Result
The test agar plates show no evidence of mycoplasma.
Notes
1. Mycoplasma colonies have a typical colony formation commonly described as “fried egg”
(See Figure 8) due to the opaque granular central zone of growth penetrating the agar
surrounded by a flat translucent peripheral zone on the surface. However in many cases
only the control zone will be visible.
2. Positive controls may be included at a concentration to give 100 colony-forming units.
These controls should obviously be handled in a laboratory remote from the main tissue
culture laboratory.
3. Control organisms (M. pneumoniae, and M. orale) are available from National Collection of
Type Cultures (UK).
4. Mycoplasma pneumoniae is a potential pathogen and must be handled in a class II
microbiological safety cabinet operating to ACDP Category 2 Conditions.
5. This test procedure should be carried out in a microbiology laboratory away from the cell
culture laboratory.
All from<<Cell Culture Cell Culture Fundamental Techniques in>>------Sigma

网友eming43的经验
支原体的检测.
PCR法
原理 该法是通过PCR技术将支原体16srRNA基因特异性扩增,来检测培养细胞中污染的支原体。PCR引物选自16sr RNA基因上的支原体特异片段,此片段与其他细菌的序列无交叉性杂交反应。扩增产物用琼脂糖凝胶电泳,经溴乙啶(EB)染色后在紫外透射仪上进行检测。
u 16sr DNA引物用水稀释至40umol/L。
(1).正向引物:5’-ACTCCTACGGGAGGCAGCAGTA-3’
(2).反向引物:5’-TGCACCATCTGTCACTCTGTTAACCTC-3’
2) PCR扩增
(1) 在冰浴中对各样品制备PCR重要混合物,对每个样品均加入:
10X PCR缓冲液 5.0ml
25mmol/L dNTPs 混合物 0.4ul
40umol/L 16sr DNA引物 每种引物1.0ul
40umol/L pBR322 DNA 引物 每种 2.0ul
pBR322 DNA 1pg/ml 2.0ul
DNA聚合酶(5U/ml) 0.2ul
三蒸水 35.4ul
总量 45ul
(2) 用无菌去离子水稀释样品为1:10,1:100。
(3) 于每个eppendorf管中加45ul PCR扩增混合物,每管中分别加入样品原液及1:10,1:100稀释液各5ul,操作均在冰浴中进行。
(4) 在每个eppendorf管中轻轻加入50ul无菌矿物油,覆盖在液面上。如DNA扩增仪带有有制式热盖,此步骤可省略。
(5) 将各管放入DNA扩增仪的孔槽内,并执行以下循环指令:
① 950C 10min 一个循环
② 950C,30S→580C 1min→720C 1min, 共30个循环。
③ 最后720C 10min延长循环。
网友bearina的意见:
在光镜下,可以看到细胞膜上吸附很多圆点,4undefined16倍下约有0.1mm,分布多集中于细胞核周围和细胞边缘,中间部位较少。严重的整个细胞都可以看到。消化时可以看到它们从细胞膜上游离出来,很多,还会动。培养基偏碱时会大量游离。感染后细胞生长变慢,凋亡率增加,正常培养时也会出现死亡。我认为是支原体污染造成的。建议培养细胞时,如发现细胞有异常,一定要高倍镜下检测。低倍镜下一般看不出细胞的变化。
gaoyunhang认为最主要是支原体污染的发现,如果发生支原体污染,细胞营养液常常变黄,细胞生长变缓,部分细胞发生脱落等等。
如果发生了支原体污染,除非的确是极为重要的细胞,最保险的方法是将被污染细胞扔掉。
所有的对支原体污染有效的药物对细胞均有一定的毒副作用,建议不要采用。
细胞培养支原体感染
??细胞培养(特别是传代细胞)被支原体污染是个世界性问题。国内外研究表明,95%以上是以下四种支原体:口腔支原体(M.orale)、精氨酸支原体(M.arginini)、猪鼻支原体(M.hyorhinis)和莱氏无胆甾原体(A.laidlawii),为牛源性。以上是最常见的污染细胞培养的支原体菌群,但能够污染细胞的支原体种类是很多的,国外调查证明,大约有二十多种支原体能污染细胞,有的细胞株可以同时污染两种以上的支原体。
??支原体污染的来源包括工作环境的污染、操作者本身的污染(一些支原体在人体是正常菌群)、培养基的污染、污染支原体的细胞造成的交叉污染、实验器材带来的污染和用来制备细胞的原始组织或器官的污染。
??组织细胞培养工作中,主要从以下几个方面来预防支原体的污染:控制环境污染;严格实验操作;细胞培养基和器材要保证无菌;在细胞培养基中加入适量的抗生素。支原体污染细胞后,特别是重要的细胞株,有必要清除支原体,常用方法有抗生素处理、抗血清处理、抗生素加抗血清和补体联合处理。支原体最突出的结构特征是没有细胞壁,一般来讲,对作用于细胞壁生物合成的抗生素,如 -内酰胺类、万古霉素等完全不敏感;对多粘菌素(polymycin)、利福平、磺胺药物普遍耐药。对支原体最有抑制活性及常用于支原体感染治疗的抗生素是四环素类、大环内酯类及一些氟喹诺酮;其他类抗生素如氨基糖苷类、氯霉素对支原体有较小抑制作用,所以常不用来作为支原体感染的化学治疗剂。InvivoGen公司研究开发的新一代支原体抗生素M-Plasmocin能有效地杀灭支原体,不影响细胞本身的代谢,并且用M-Plasmocin处理过的培养细胞,不会重新感染支原体。
1、支原体肺炎的首选药是红霉素和四环素(副作用多,已少用)。所以用红霉素是有道理的。但是,由于耐药菌的泛滥,红霉素耐药菌非常多,所以有人发现红霉素无效(当然可能根本不是支原体污染),要不可以试试四环素,用的人少,耐药菌还是要少一些的,然而,红霉素和四环素都是抑菌剂,所以不能断根,这就是我下面要谈的。
2、对支原体有效的杀菌剂还是氨基甙类,即使只有中度敏感。这里面就有一个问题,很多人又用庆大霉素又用卡那霉素,我不知道这是为什么?他为什么不再加上链霉素和妥布霉素?协同作用是由不同机制的药物合用产生的,同一机制药物的作用仅等同于增加其中某种药物的剂量而已,有什么意义呢?所以我觉得用一种就够了。庆大霉素不错,但耐药菌也常见,所以有时可能还是没效果。丁胺卡那是这类药中较少产生耐药性的,可以试试吗?
3、谈谈合用的问题。我推荐抑菌剂+杀菌剂:红霉素/四环素+庆大霉素/丁胺卡那霉素,我个人认为四环素+丁卡可能最为有效,因为相对而言,细菌对他们都教陌生、敏感。但是还不够,请往下看。
4、谁都不敢保证这样就万事大吉了。里面有一小撮细菌可能还能顽强抗过去,它们的后代必将变异为耐药菌。所以还应该加点其他的措施,因为这些细菌量毕竟少,消化后的悬浮细胞离心一下,细胞毕竟比支原体重,是不是可以分离出来?多次稀释细胞并离心是否可以很大程度的减少支原体的数量呢?当然,前提是用了一到两次抗生素细菌只有少量了。
5、再不行,复苏吧。
我们实验室所有细胞都有支原体污染,而且从上个学期以来越来越严重了。本来上个学期细胞生长还比较快,而实验室负责细胞房管理和复苏细胞、配培基的老师看了以后也总说没关系,因此一直没在这事上下工夫,细胞长得太糟我就用D-Hanks洗两次,然后用酶消化后用灭菌的离心管洗细胞、1000rpm离心,三次,同时用D-Hanks洗两次培养瓶,重新贴壁,似乎也有一点点效果。但这个学期要用的几种细胞中有的过了两个星期也长不起来,长起来的细胞周围也有很多沙子样的小黑点,全实验室所有养了细胞的都说自己的细胞长得差,负责细胞房工作的老师复苏了一次又一次,这方面的问题还是没有人着手解决,我急了,查了支原体方面的文献,有些文章比较麻烦,注射到小鼠体内来杀灭支原体,至少这在我们实验室是不可能使用的方法;有些文章是联合使用两种抗生素来清除支原体,简单方便,我就试了这种。
关于检验支原体污染,有PCR法、培养法、DNA荧光染色法,我选择的是DNA荧光染色法,将冰醋酸和甲醇按1:3混合,固定细胞两次,每次10分钟,然后用1μg/ml的Hoechst33258染色20分钟,紫外激发,确认了我的细胞中生长最好的都有支原体污染。于是我花1.5元买了环丙沙星(ciprofloxacin)药片,稀释成50μg/ml,离心去除淀粉之类的不溶性辅料,加入培基中,稀释成10μg/ml,跟正常培基一样用法,打算处理12天后再用Hoechst33258染色验证,对于污染严重的再用1μg/ml四环素处理3天。我前两天试的,每种细胞分成两瓶,一瓶用加ciprofloxacin的培基,一瓶不加,对比明显,效果不错,但要完全消除还需要一些天。我向同学推荐了这个方法,有人说自己的培基前段时间已经加了青霉素,但好象没有效果,支原体没有细胞壁,这类干扰细胞壁合成的抗生素当然对其没有效果;有的同学试了,细胞变得漂亮多了,原来牢牢贴在瓶壁上的黑点也去掉了。呵呵,俺的那些细胞现在是越长越标致了。
下图是支原体污染细胞的Hoechst33258染色结果。

细胞一旦污染支原体,留之又不能,弃之又可惜,所以总要想一些补救措施,现将我所知道的与各位交流一下。
1)化学药物——在上面讲了很多了
2)抗血清处理——特异性多抗
3)6-甲基嘌呤脱氧核敢(6-MPDR)有限稀释法
4)激活巨噬细胞的应用——可在24孔板内每孔加入1ml饲养细胞20万/ml,使形成巨噬细胞单层,再加入所培养的细胞。巨噬细胞培养基中加入四环素、红霉素、tylosin或林可霉素等抗生素,可消除污染
5)克隆法——有限稀释法,是将细胞稀释接种96孔板中,使每孔接种一个细胞,挑取无污染的单个克隆,此法对污染较轻的细胞株有比较好的效果。
6)加热法——利用软琼脂技术,50度加热灭活细胞的支原体,再于37度1-3天培养,使琼脂中的抗生素与支原体进一步作用。
7)小鼠体内传代除去支原体污染——被支原体污染的特异性杂交瘤细胞可接种鼠腹腔中传代,待长出肿瘤后取出接种小鼠,如此反复2-3次,可有效清除。这种方法在实际单抗制备杂交瘤污染时特别适用。
zxcvbmumu试过了,使用终浓度为10ug/ml的乳酸环丙沙星注射液,可以抑制细胞间的黑点,并使细胞恢复增殖活性,但是好像不能完全去除黑点,谢谢前面讲这一方法的所有发帖者!