生物实验新手上路之GATEWAY技术
互联网
- 相关专题
GATEWAY技术可将基因转入多个表达系统并大大节省您的时间,是近年来令人瞩目的克隆 表达新方法。现特收集gateway技术的资料供大家学习。(更多阅读克隆 、表达新方法之Gateway技术 )
gateway技术近两年开辟了高效克隆转载新方法,Gateway克隆技术是利用λ噬菌体与大肠杆菌的染色体之间发生的位点特异性的重组 整合(integrative recombination reaction)与切出(excisive recombination reaction)反应。
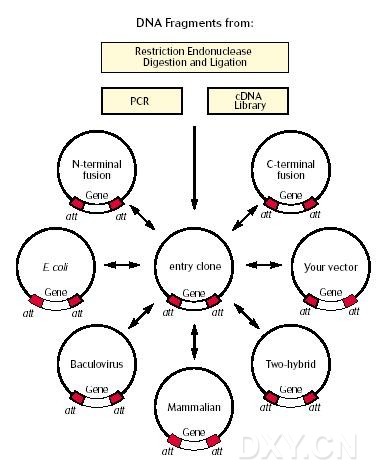
GATEWAYTM克隆 中基因看上去是什么样的?
基因两端应该有att位点,在Entry克隆 中是att L序列(100bp),而在表达克隆中是att B序列(25bp)[Hartley,
J. L., Temple, G. F., 和Brasch, M. A. (2000) Gen. Res.
101788.]。在att位点间的所有序列会和你的基因一起移动。因此,你必须预先决定是否包含诸如真核或原核翻译信号,或3'终止编码等DNA元件。
我能克隆多大的片段?
对于PCR产物,我们已经克隆从100bp到10kb大小片段的同时,LR反应已经用于操作130kb大小的目标载体。
最小的片段是什么?
我们相应必定有一个最小尺寸,可以是由于重组反应的拓扑结构所限制的。早期数据显示,位点间有7bp片段就足够了。
我怎样利用这一系统?
PCR是最容易的方法。扩增你的基因,在其N-末端含有attB1位点,C-末端含有attB2位点。网站上有链接到GATEWAY信息,你通过点击按键即可在你的引物上按顺序加上att位点。你可以利用限制性内切酶和连接酶将你的基因克隆到Entry
Vectors中或者购买已包含attB载体的cDNA文库(PCMV.SPORT6或PSPORT-P)。
为什么你们推荐在PCR引物中加上attB位点并且转化PCR产物到一个供体载体中,然后被重组(与attL)进attR位点?为什么PCR引物不能包含attL位点以便直接重组到目标载体中而不需要Entry
Clone的中介呢?
Entry Clone是作为基因的一个来源克隆以便较容易地亚克隆到任意目标载体。同时,一旦Entry
Clone是测序有效的,就不需要对多个表达载体进行测序。另外,attB序列相对较小(25bp),这使得它们更适合加到PCR引物中去(需要在5'端附加4个G以便进行有效重组)。attL识别序列是100bp,attR是125bp,而attp是233bp。
一个PCR产物能否直接克隆到目标载体中?
可以使用一管法。
步骤1:进行BP反应将PCR产物克隆到pEONR201中。
步骤2:加入试剂进行LR反应。因此,可以1天中在1个反应管中进行BP和LR反应。然后,转化反应产物并直接分离你的表达克隆。
通过PCR加入attB位点需要什么纯度的引物?
标准纯度对许多PCR是足够的,我们发现用HPLC纯化的标准GATEWAY引物(<50碱基)与标准纯度相比可增加2倍的克隆数目。如果你得到很少的克隆,你可以增加PCR产物用量,或增加BP反应的保温时间,或两者兼顾以提高克隆数目,对大于5kb的PCR产物并且大于65碱基的引物,推荐使用纯化的引物(如Cartridge,HPLC和PAGE)以增加克隆数目。
我能将我的cDNA文库转到GATEWAY载体中以便产生一个GATEWAY文库吗?
这并不容易。你可以用包括cDNA并包含attB位点的引物进行PCR,但使用PCR是有倾向性的。如果你用限制性内切酶来移动它,任何内部含有该酶切位点的cDNA也会被消化。最好的方法是在一个包含attB的载体中重建该文库。
增加BP反应时间会改善效率吗?
是的,将反应时间由1小时增加到4小时会增加2到3倍的克隆数目。过夜反应则会增加5到10倍。
起始的ATG位点与5'attB位点的相对位置如何?
这取决于你是想表达一个融合或一个天然蛋白。对于一个N-末端融合蛋白,ATG位点会位于attb1位点的上游(在目标载体中)。而对于一个天然蛋白的表达,会在attB1位点下游找到ATG位点。
att位点会影响蛋白的表达吗?
即使attB位点将8个或9个氨基酸加到融合蛋白中去,我们也没有观察到att位点影响蛋白的表达或稳定性的情况。在大肠杆菌,昆虫或是哺乳动物细胞或是在酵母中的双杂交反应中,attB1编码的氨基酸在融合蛋白的氨基末端都没观察到对蛋白表达产生影响。同样,研究人员[Simpson,
J, C., Wellenreuther, R., Poustka A., Pepperkok, R.和Wiemann, S. (2000)
EMBO. Reports, 1:287]显示用GATEWAY技术克隆的基因表达的蛋白在细胞中的定位可以得到保持。
我如何将一个分泌型前导序列编入Enrty载体中?
一个简单的方法来表达含有前导序列的蛋白是让目标载体编码前导序列。否则你将不得不利用好限制性内切酶将前导序列亚克隆到Entry载体中。
我能用什么限制性内切酶将我的目标载体线性化?
可以使用能在GATEWAY盒中切割的酶,但要注意避开ccdB基因。所用的GIBCOBRLTM目标载体都是以线性状态提供的。当没有合适的单切点的酶切位点可以利用或不确定时,可以用拓扑异构酶简单处理以线性化DNA分子(参见说明书中的改进方法)。
用于GATEWAY反应的DNA必须什么样的纯度?
小量制备(碱裂解法)的DNA即可用于在GATEWAY反应。然而,通常,这种DNA分子由于含有RNA和核酸而难以通过紫外分光光度计来定量。推荐使用CONCERTTMDNA纯化系统提纯的质粒DNA。
对于PCR片段,推荐使用PEG沉淀或凝胶纯化并用CONCERT快速凝胶抽提系统进一步纯化。
不正确重组的频率有多高?
因为GATEWAY克隆是基于λ位点特异重组,不会发生不正确的重组现象。
个人总结
优点:
1. 效率高,空载体含有毒性基因所以避免了大部分的假阳性。
2. 在不同载体之间转换方便,省去了大量的酶切/胶回收/去磷酸/连接的步骤,尤其适合筛选大量基因和突变体的应用。
3. 速度快,大部分反应1小时完成而且转化效率很高,自制的感受态DH10B(1ug=10^8cfu)加1ul反应产物就可以。
4. 反应缓冲液是TE,可以直接电转化
5. 兼容TOPO,可以用CmR/ccdB片断构建载体
6. 个人感觉容错率较高,对时间和温度的控制要求也不严。一开始没经验,用水浴加温,拿出来发现一个EP管里的液量神奇的增加了,应该是进了水。硬着头皮继续做居然成功了。
缺点:
1. 酶很贵,真的很贵
2. 很贵的酶还很娇贵,冰上几分钟就有可能失活。虽然现行版本的酶经过改进只需要零下20度但是为了以防万一还是推荐零下80度保存。
3. 10kb以上的片断需要隔夜培育,和传统的酶切-T4连接相比没有优势
4. 载体选择比前几年多但是依然较少,invitrogen的质粒很贵,自己构建和扩增载体较繁琐而且需要抗ccdB的特殊菌株来转化。
下面说一些手册里没有的小技巧
1. 手册里的反应体系是10ul, 其实完全可以减半, 不影响效果。2.5ul的体系试过但是因为体积太小所以可重复性较差,不推荐。手册里的一步反应法也很奢侈,实际操作中BP反应产物直接取2ul加入LR反应体系即可。
2. BP反应试剂盒里的阳性控制不推荐,一来检测麻烦(需要四环素平板),二来没有必要还浪费酶。BP反应后可以取1ul产物用M13测序引物做PCR然后跑胶。正常情况下应该能扩出两个条带: 1.7kb的一条是空白载体上的cmR和ccdB片断,而另一段的长度应该等同于Attb片断的长度。反应完全的情况下两个条带的亮度应该差不多(BP反应是可逆的)。如果目的基因的条带很暗的话可以适当增加转化用的产物量或者选用好一些的感受态菌株。
3. PCR反应要先纯化否则残余引物会影响反应。BP试剂盒里的PCR纯化剂(PEG8000/MgCL2)效果不错但是离心后很难看到DNA沉淀,只能按照感觉吸去上清然后加入TE测定核酸含量判断有没有回收成功。普通的硅胶离心柱纯化法贵一些,但是相对可靠。
4. 手册里说BP反应里的AttB片断必须是线性的,如果用质粒必须做酶切,但是事实证明用超螺旋的质粒结果没有区别。
5. 同理,用引物在目的基因上下游加入AttL序列后直接进行LR反应也是可行的,可以省去BP反应相关的材料和费用。但是pDONR系列载体自有其优点(载体小,拷贝数高,卡那霉素抗性,容易测序),所以要按实际情况权衡
6. 慢病毒载体的空白质粒有大量的重复序列,久存会导致LR反应成功但是转化后抽不出质粒的情况,需要注意。
Gateway官方简介:
Gateway®克隆技术的基础是lambda噬菌体的位点特异性重组反应,一种准确保存遗传信息的有效的自然的生化途径。与传统的需要DNA限制性内切酶、DNA连接酶、凝胶电泳和DNA片段纯化等多个步骤的单调乏味的亚克隆方法相比,该方法只需一步生化反应而且操作方便、快捷(室温60分钟),节省了大量的时间和劳力。另外,根据目的蛋白的生产和纯化的不同要求, Invitrogen公司提供一系列含有不同启动子和融合标签、以及在几种宿主表达的目标载体。
最后提醒各位,其实Gateway克隆技术还是挺好上手的,平常记得多翻翻说明多看看文献,再多总结经验,就OK了。