PCR引物设计之个人心得篇
互联网
|
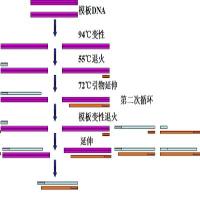
记得当初写本科论文,感到不知道讨论什么问题好。愣是写了一大段的PCR条件摸索的讨论。后来PCR成为实验最基本的一步了,但是发现在PCR中还是有许多需要注意的地方。PCR的第一步就是引物设计了。 引物设计需要注意的地方很多,在大多数情况下,我们都是在知道已知模板序列时进行PCR扩增的。在某些情况比如构建文库的时候也会在不知道模板序列的情况下进行设计。这个时候随机核苷酸序列就与模板不是完全匹配。 我们通常指的设计引物都是在已知模板序列的情况下进行。 设计的目的是在两个目标间取得平衡:扩增特异性和扩增效率。引物分析软件将试图通过使用每一引物设计变化的预定值在这两个目标间取得平衡。 设计引用有一些需要注意的基本原理:
在引物长度小于20bp时:[4(G+C)+2(A+T)]-5℃ 在引物长度大于20bp时:62.3℃+0.41℃(%G-C)-500/length-5℃ 另外有许多软件也可以对退火温度进行计算,其计算原理会各有不同,因此有时计算出的数值可能会有少量差距。为了优化PCR反应,使用确保退火温度不低于54℃的最短的引物可获得最好的效率和特异性。 总的说来,每增加一个核苷酸引物特异性提高4倍,这样,大多数应用的最短引物长度为18个核苷酸。引物长度的上限并不很重要,主要与反应效率有关。由于熵的原因,引物越长,它退火结合到靶DNA上形成供DNA聚合酶结合的稳定双链模板的速率越小。 ② GC含量
③ 退火温度
④ 避免扩增模板的二级结构区域
⑤ 与靶DNA的错配
最后在3处点击Align就可以查看引物在靶DNA中是否有多个同源位点了。 可是使用BLAST还是有其不方便的地方。因为它一次只能比较两条序列,那么一对引物就需要分开进行比对。如果存在错配,还需要自己计算由于错配形成的片段长度有多大。在下一篇中将介绍一个软件,可以直接将靶DNA和引物输入对产物片段进行预测。 ⑥ 引物末端
3′端也不能有形成任何二级结构可能,除在特殊的PCR(AS-PCR)反应中,引物3′端不能发生错配。如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增特异性与效率。 ⑦ 引物的二级结构
⑧ 为了下一步操作而产生的不完全匹配
很多时候PCR只是初步克隆,之后我们还需要将目的片段亚克隆到各种载体上,那么就需要在PCR这个步骤为下一步的操作设计额外的碱基。以下总结一些为了亚克隆所要设计的序列。 a 添加限制性内切酶酶切位点
但是不同的酶需要的保护碱基数目是不相同的,例如:SalⅠ不需要保护碱基,EcoRⅤ需要1个,NotⅠ需要2个,Hind Ⅲ 3个。其中,在原核表达设计引物时还有一些小技巧,大家可以参考:《原核表达之实验前的分析》。里面一些规则是所有表达都通用的。 有一种做法是在进行PCR反应的同时进行酶切,这样就需要注意一些内切酶在PCR反应中的酶切反应率,见附录。不过这种方法虽然方便但并不推荐。有时候,就是把PCR产物回收后酶切再与载体连接效果都不尽理想,同步进行会使出现问题的原因变得更加复杂。一旦出现问题,分析起来更麻烦。 b LIC添加尾巴
扩增目的插入片段的引物5'序列要与LIC载体互补。T4 DNA 聚合酶的3'→5'外切活性经短时间即可在插入片段上形成单链粘端。由于只能由制备好的插入片段和载体互相退火形成产物,这种方法非常快速高效,而且为定向克隆。 c 定向TA克隆添加尾巴
d In-Fusion克隆方法
只要在设计引物的时候引入一段线性化载体两端的序列,然后将PCR产物和线性化的载体加入到含有BSA的In-Fusion酶溶液中,在室温下放置半个小时就可以进行转化了。这种方法特别适合大批量的转化。 这里顺便提一下如果有什么技术给大家留下深刻影响,欢迎大家发email推荐给生物通。说不定你的推荐可以让它成为年度之星呢。 如果要加入额外的碱基总是或多或少会影响到整个PCR反应,比如在加入NotⅠ的酶切位点后整个引物的退火温度就会直线上升(它识别的是8个碱基,且全为GC),这样使另外一个引物的设计变得十分困难,因为一对引物间退火温度相差不宜太远。 因此上面提到许多设计原则在实际应用中往往难以做到都符合。在碰到这些情况的时候,我们只能秉着“实践是检验真理的唯一标准”这一原则,要试一试才能知道能否行得通了。 |