实时荧光定量PCR原理和实验
互联网
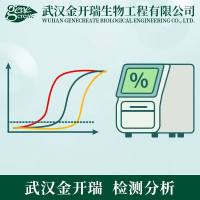
无论是对遗传病(如地中海贫血和血友病)、传染病(如肝炎和艾滋病)或肿瘤进行基因诊断,还是研究药物对基因表达水平的影响,或者监控药物和疗法的治疗效果,定量PCR技术都可以发挥很大作用。定量PCR技术的最新进展是实时荧光定量。该技术借助于荧光信号来检测PCR产物,一方面提高了灵敏度,另一方面还可以做到PCR每循环一次就收集一个数据,建立实时扩增曲线,准确地确定CT值,从而根据CT值确定起始DNA拷贝数,做到了真正意义上的DNA定量。这是DNA定量技术的一次飞跃。 根据最终得到的数据不同,定量PCR可以分为相对定量和绝对定量两种。典型的相对定量如比较经过不同方式处理的两个样本中基因表达水平的高低变化,得到的结果是百分比;绝对定量则需要使用标准曲线确定样本中基因的拷贝数或浓度。根据所使用的技术不同,荧光定量PCR又可以分为TaqMan探针和SYBR Green I荧光染料两种方法。比较而言,探针杂交技术在原理上更为严格,所得数据更为精确;荧光染料技术则成本更为低廉,实验设计更为简便。在选择实验方案时要根据实验目的和对数据精度的要求来决定。 定量实验与定性实验最大的不同,是要考虑统计学要求并对数据进行严格的校正,以消除偶然误差。因此重复实验和设立内对照非常重要。由于各种各样的客观原因,这一点在实践中往往被轻视或忽视,需要着重强调。当然,与定性实验一样,定量PCR也要设立阴性和阳性对照,以监控试剂和实验操作方面可能出现的问题。 1. 为什么终点定量不准确? 我们都知道理论上PCR是一个指数增长的过程,但是实际的PCR扩增曲线并不是标准的指数曲线,而是S形曲线。这是因为随着PCR循环的增多,扩增规模迅速增大,Taq酶、dNTP、引物,甚至DNA模板等各种PCR要素逐渐不敷需求,PCR的效率越来越低,产物增长的速度就逐渐减缓。当所有的Taq酶都被饱和以后,PCR就进入了平台期。由于各种环境因素的复杂相互作用,不同的PCR反应体系进入平台期的时机和平台期的高低都有很大变化,难以精确控制。所以,即使是重复实验,各种条件基本一致,最后得到的DNA拷贝数也是完全不一样的,波动很大。 传统的定量方法,如溴乙锭染色或放射性核素掺入标记后的光密度扫描等,测定的都是PCR的终产物,而不是起始DNA拷贝数。由于PCR的终产物量与起始模板量之间没有线性关系,所以根据最终的PCR产物量不能计算出起始DNA拷贝数。 对于绝大多数实验,比如甲肝的诊断、药物疗效的监测等,需要测定的都是PCR放大之前标本中的DNA原始拷贝数,经过PCR扩增以后的DNA拷贝数已经不能反映真实情况。在这种情况下,就不能采用终点定量,而要根据CT值确定DNA起始拷贝的数量。 2. 为什么CT值与起始模板拷贝数成线性关系? CT值的定义是PCR扩增过程中,荧光信号开始由本底进入指数增长阶段的拐点所对应的循环次数。从图1的重复实验中可以直观地看到,尽管平台期DNA拷贝数波动很大,CT值却是相对固定的。如果用不同浓度的DNA作PCR,可以看出DNA浓度越高,CT值越小。DNA浓度每增加1倍,CT值减小1个循环。CT值与模板DNA的起始拷贝数成反比。 这一结论可以从数学上严格证明。为使表达式简便,以下推导忽略PCR效率等细节。如果考虑这些因素,可以在方程上增加修正项。这些修正项的增加并不改变方程的线性性质。 一般地,我们有Rn=RB+XO(1+EX)nRS,也就是说第n次PCR循环时的荧光信号强度(Rn)等于背景信号强度(RB)加上每个分子的荧光强度(即单位荧光强度,RS)与分子数目的乘积。当循环次数n = CT时,则有RT=RB+XO(1+EX)CTRS。两边取对数,得log(RT -RB) = logX0 + CTlog(1+ Ex) + logRs。整理此式,CTlog(1+ Ex) = - logX0 + log(RT -RB)–logRs。 所以对于每一个特定的PCR反应来说,EX、RT、RB和RS都是常数,所以CT值与log X0成反比,也就是说,CT值与起始模板拷贝数(X0)的对数成反比,起始DNA浓度每增加1倍,CT值减小1个循环。根据CT值的定量是精确和严格的,而传统的终点定量则比较粗放。 如果读者有兴趣的话,也可以假设PCR的效率(即Ex)为100%,从上式推算出定量PCR标准曲线的最佳斜率和CT值的最佳范围。 3. 怎样确定CT值? 实验操作中,CT值定义为在基线上方产生可检测到的统计学上显著的荧光发射时所对应的PCR循环次数。“基线上方”也就是阈值高度的量化定义是基线范围内荧光信号强度标准偏差的10倍。阈值所在的横线与PCR扩增曲线的交点所指的PCR循环次数就是CT值。基线范围的定义是从第3个循环起到CT值前3个循环止,其终点要根据每次实验的具体数据调整,一般取第3到第15个循环之间。早于3个循环时,荧光信号很弱,扣除背景后的校正信号往往波动比较大,不是真正的基线高度;而在CT值前3个循环之内,大多数情况下荧光信号已经开始增强,超过了基线高度,都不宜当作基线来处理。
显然,CT值取决于阈值,阈值取决于基线,基线取决于实验的质量,CT值是一个完全客观的参数。CT值越小,模板DNA的起始拷贝数越多;CT值越大,模板DNA的起始拷贝数越少。正常的CT值范围在18-30之间,过大和过小都将影响实验数据的精度。 4. 荧光信号和定量数据的归一化 虽然大多数定量PCR仪都会自动扣除本底,但是仍然需要注意荧光信号强度和定量数据的归一化校正,以便不同样本之间的实验数据可以严格地相互比较。 由于加样操作的误差、离心管透光性能的差异、荧光激发效率的差异等偶然误差不可避免,因此仪器收集到的原始信号必须进行归一化校正,以消除这些因素对定量结果的影响。这种校正可以通过在反应体系中添加额外的荧光染料来实现,一般采用红色ROX荧光,称为阳性参比信号。ROX在反应缓冲液中的浓度是固定的,因此其信号的强度与反应体系的总体积和总的荧光激发效率正相关。目标基因和对照基因的信号除以阳性参比信号以后,即Rn = R / RROX,就可以在同样的起点上进行比较和各种计算。 ROX校正可以提高定量数据的精度和重现性,减少孔间差异。 归一后的荧光信号再扣除本底,就得到DRn:DRn = Rn,样本- Rn,空白。DRn是最后构建PCR实时扩增曲线的纵坐标。 无论绝对定量还是相对定量,在得到实验结果后,还要考虑数据之间的可比性问题。在实验操作中,取样都是以体积或重量为单位的,但是同样体积或重量的样本所来源的细胞数目并不一样,所以拷贝/μL或拷贝/ng的定量数据相互之间实际上并不可比。只有将这些数据归一到以拷贝/细胞或拷贝/基因组为单位后,才可进行严格意义上的比较。 这种校正可以通过适当的参比来完成。参比一般选用b-actin、GAPDH、rRNA基因等管家基因。由于它们在细胞中的表达量或在基因组中的拷贝数是恒定的,受环境因素影响较小,其定量结果代表了样本中所含细胞或基因组的数量。校正方法为:[DNA]样本/ [DNA]IPC,或者DCT = CT,样本- CT, IPC。因为CT值与起始DNA拷贝数的对数是反比关系,可以证明,这两种计算方法在数学上是等价的。 为了减少误差,目标基因和参比基因最好在同一反应管内同时进行定量测定,所以这种对照称为阳性内对照(Internal Positive Control,IPC)。要进行IPC归一化校正,定量PCR仪必须具备多色检测能力,最好是4色。否则,目标基因和参比基因只能分两管作定量,就不成其为“内”标了。 5. 污染的预防和热启动 为保证定量的准确性,要预防非特异性PCR扩增和污染。常用的措施有使用UNG酶(Uracil-N-Glycosylase)和热启动。UNG酶的作用原理是降解含有dU的双链或单链DNA。它在50°C激活,95°C灭活。由于商用PCR试剂盒均以dUTP取代dTTP,所以PCR产物都是含有dU的DNA链。在定量PCR开始前增加50°C的保温步骤,UNG酶即可将已有的PCR产物降解破坏,防止可能造成的污染。 普通的Taq酶即使在室温下也有一定的活性,如果不采取措施,在加入PCR试剂的过程中、正式PCR开始前就会完成少量PCR扩增,增加了背景,影响定量精度。而金牌Taq酶经过特殊修饰,常温下其活性部位被封闭,没有活性;只有经过95°C 10 min的热启动以后,封闭被解除,才能开始DNA链延伸,这样就最大限度地减少了杂讯的生成。 6. 标准曲线、重复实验和阴性、阳性对照 定量实验,误差是不可避免的。设立重复实验,对数据进行统计处理,可以将误差降低到最小。所以定量实验的每个样本至少要重复3次以上,严格的定量更应当重复6~8次,以满足小样本统计的要求。 如果作绝对定量,则标准曲线需要在5个点以上。标准曲线使用的标准品是浓度已知的DNA样本,可以自己制备,也可以购买商品化的试剂盒。其PCR反应条件应当与未知样本的一致,以便在同一反应板上同时定量。 阴性对照中不加模板DNA,而以水或缓冲液代替,用于检验是否存在PCR污染。阳性对照则用于检验PCR试剂和实验操作上可能出现的问题。如果实验中设立了IPC,则IPC既可以用来校正数据,也可以起到阳性对照的作用。 7. TaqMan探针技术原理 TaqMan探针法是高度特异的定量PCR技术,其核心是利用Taq酶的3′→5′外切核酸酶活性,切断探针,产生荧光信号。由于探针与模板是特异性结合,所以荧光信号的强弱就代表了模板的数量。 在TaqMan探针法的定量PCR反应体系中,包括一对PCR引物和一条探针。探针只与模板特异性地结合,其结合位点在两条引物之间。探针的5′端标记有报告基团(Reporter, R),如FAM、VIC等,3′端标记有荧光淬灭基团(Quencher, Q),如TAMRA等。当探针完整的时候,报告基团所发射的荧光能量被淬灭基团吸收,仪器检测不到信号。随着PCR的进行,Taq酶在链延伸过程中遇到与模板结合的探针,其3′→5′外切核酸酶活性就会将探针切断,报告基团远离淬灭基团,其能量不能被吸收,即产生荧光信号。所以,每经过一个PCR循环,荧光信号也和目的片段一样,有一个同步指数增长的过程。信号的强度就代表了模板DNA的拷贝数。 TaqMan探针根据其3′端标记的荧光淬灭基团的不同分为两种:普通的TaqMan探针和TaqMan MGB探针。MGB探针的淬灭基团采用非荧光淬灭基团(Non-Fluorescent Quencher),本身不产生荧光,可以大大降低本底信号的强度。同时探针上还连接有MGB (Minor Groove Binder)修饰基团,可以将探针的Tm值提高10°C左右。因此为了获得同样的Tm值,MGB探针可以比普通TaqMan探针设计得更短,既降低了合成成本,也使得探针设计的成功率大为提高。因为在模板的DNA碱基组成不理想的情况下,短的探针比长的更容易设计。实验证明,TaqMan MGB探针对于富含A/T的模板可以区分得更为理想。 8. SYBR Green I 荧光染料技术原理 SYBR Green I是一种只与DNA双链结合的荧光染料。当它与DNA双链结合时,发出荧光;从DNA双链上释放出来时,荧光信号急剧减弱。因此,在一个体系内,其信号强度代表了双链DNA分子的数量。SYBR Green荧光染料法定量PCR的基本过程是:1、开始反应,当SYBR Green染料与DNA双链结合时发出荧光。2、DNA变性时,SYBR Green染料释放出来,荧光急剧减少。3、在聚合延伸过程中,引物退火并形成PCR产物。4、聚合完成后,SYBR Green染料与双链产物结合,定量PCR系统检测到荧光的净增量加大。 SYBR Green I荧光染料能与所有的DNA双链相结合,对DNA模板没有选择性,所以特异性不如TaqMan探针。要想用荧光染料法得到比较好的定量结果,对PCR引物设计的特异性和PCR反应的质量要求就比较高。在此前提下,本法是一种成本低廉的选择。 |