手把手教你用 Fast Tree 快速构建序列进化树
丁香园
常见的建树方法有:
贝叶斯法(Bayesian),最大似然法(Maximum likelihood,ML),最大简约法(Maximum parsimony,MP),邻接法(Neighbor-Joining,NJ),最小进化法(Minimum Evolution,ME),类平均法(UPGMA)。
一般来讲,如果模型合适,最大似然法的效果较好。对于近缘序列,最大简约法用的假设最少,各种方法结果相似。而对于远缘序列,一般使用最大似然法或邻接法。对相似度很低的序列,邻接法往往出现 Long-branch attraction(LBA,长枝吸引现象),严重干扰进化树的构建。对于各种方法构建分子进化树的准确性,Hall 认为贝叶斯的方法最好,其次是最大似然法,然后是最大简约法。其实如果序列的相似性较高,各种方法结果差别不大。
最大似然法和邻接法需要选择模型。对于蛋白质序列,一般选择 Poisson Correction(泊松修正)模型。而对于核酸序列,一般选择 Kimura 2-parameter(Kimura-2 参数)模型。
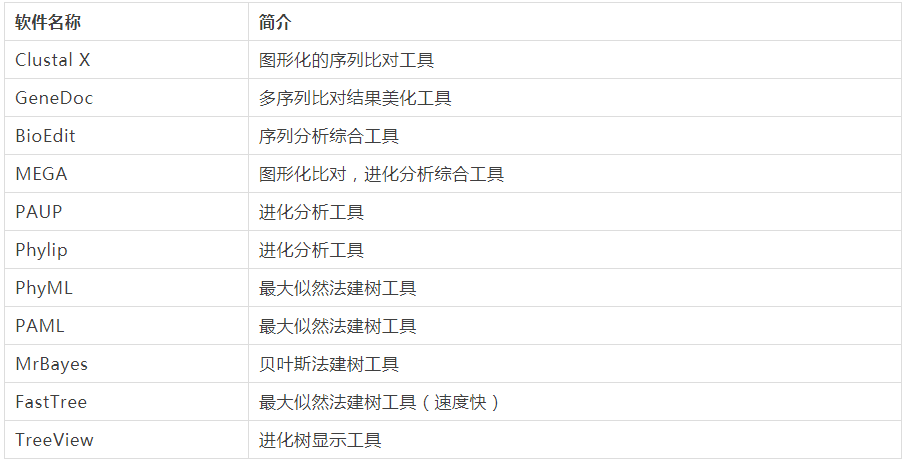
表 1. 构建进化树的常用软件
软件名称 | 简介 |
Clustal X | 图形化的序列比对工具 |
GeneDoc | 多序列比对结果美化工具 |
BioEdit | 序列分析综合工具 |
MEGA | 图形化比对,进化分析综合工具 |
PAUP | 进化分析工具 |
Phylip | 进化分析工具 |
PhyML | 最大似然法建树工具 |
PAML | 最大似然法建树工具 |
MrBayes | 贝叶斯法建树工具 |
FastTree | 最大似然法建树工具(速度快) |
TreeView | 进化树显示工具 |
本文主要讲 FastTree 使用方法:
首先介绍几点特性:
1. 在默认参数下,FastTree 比 PhyML 更准确,比 PhyML 快 100~1000 倍;
2. FastTree 使用模型为:核酸进化模型:Jukes-Cantor 或者 GTR(generalized time-reversible);蛋白进化模型:JTT (Jones-Taylor-Thornton 1992), WAG (Whelan & Goldman 2001) 或者 LG (Le and Gascuel 2008)
下载,安装 FastTree
FastTree 提供以下几个版本:
· Linux 64-bit executable (+SSE)
· Multi-threaded executable (+SSE +OpenMP) (see usage guide)
· Windows 32-bit command-line executable (no SSE)
· C code
下载 Windows 32-bit command-line executable (no SSE) 后,是一个 FastTree.exe 文件,可以直接在 cmd 命令行程序中调用运行。
新建一个文件夹:比如在 D 盘目录下新建一个 FastTree 文件夹,将 FastTree.exe 程序放在 D:FastTree 目录下。
FastTree 运行(Windows 为例)
· 开始菜单—搜索—cmd
· 切换目录到 D:FastTree
· 最大似然树构建:FastTree protein alignment file > tree
· 在目录 D:FastTree 生成. tree 文件,可以使用 TreeView 或 MEGA 打开。
· 构建进化树时,可以选择不同的模型:
命令行:D:FastTree>FastTree -lg CIPK.phy >CIPK.tree
alignment file 格式
alignment file 格式如上图。
可以首先使用 Clustal X 比对序列:Alignment—Output Format Options—Phylip format
比对后,在比对目录下生成几个文件,其中. phy 后缀名文件是 FastTree 要使用的。