


想研究基因的进化压力分析?强推这个免费易学的网站!
丁香园
遗传学和基因组学方法的快速发展丰富了我们对自然选择和分子进化的理解。基因组内含有适应性突变的基因在正向自然选择中因为占据优势而不断增加,而通过比较不同群体之间的核苷酸序列可以识别这些基因,对进化压力的检测和量化是近年来研究的一个热门领域,这些带适应性突变的基因是一般也是药物的潜在靶点。
今天小编给大家安利一个专门分析进化压力的网站:
Datamonkey
Datamonkey 进化压力在线分析平台可以识别基因中受到正面或负面选择影响的位点,并且 Datamonkey 采用纯交互视图界面,比同类命令行软件如 PAML 更加简洁,使用起来更加方便友好。
快速可靠的统计方法对于实现复杂的大规模序列进化分析是至关重要的。Datamonkey 嵌套的 HyPhy 软件包提供了几种常用的识别蛋白质编码区域中快速进化(正向选择)以及异常(负向选择)保守的位点的统计方法,通过计算位点特异性同义 (dS) 和非同义 (dN) 取代率 ω(即 dN/dS)以及统计检验来推断基因中自然选择的强度。
打开 Datamonkey 主页,会有提示引导你根据分析的要求(检测选择压力或重组、全局的或者局部的、位点特异的或者分支特异的、是大数据集或是小数据集等),选择相应的分析方法。
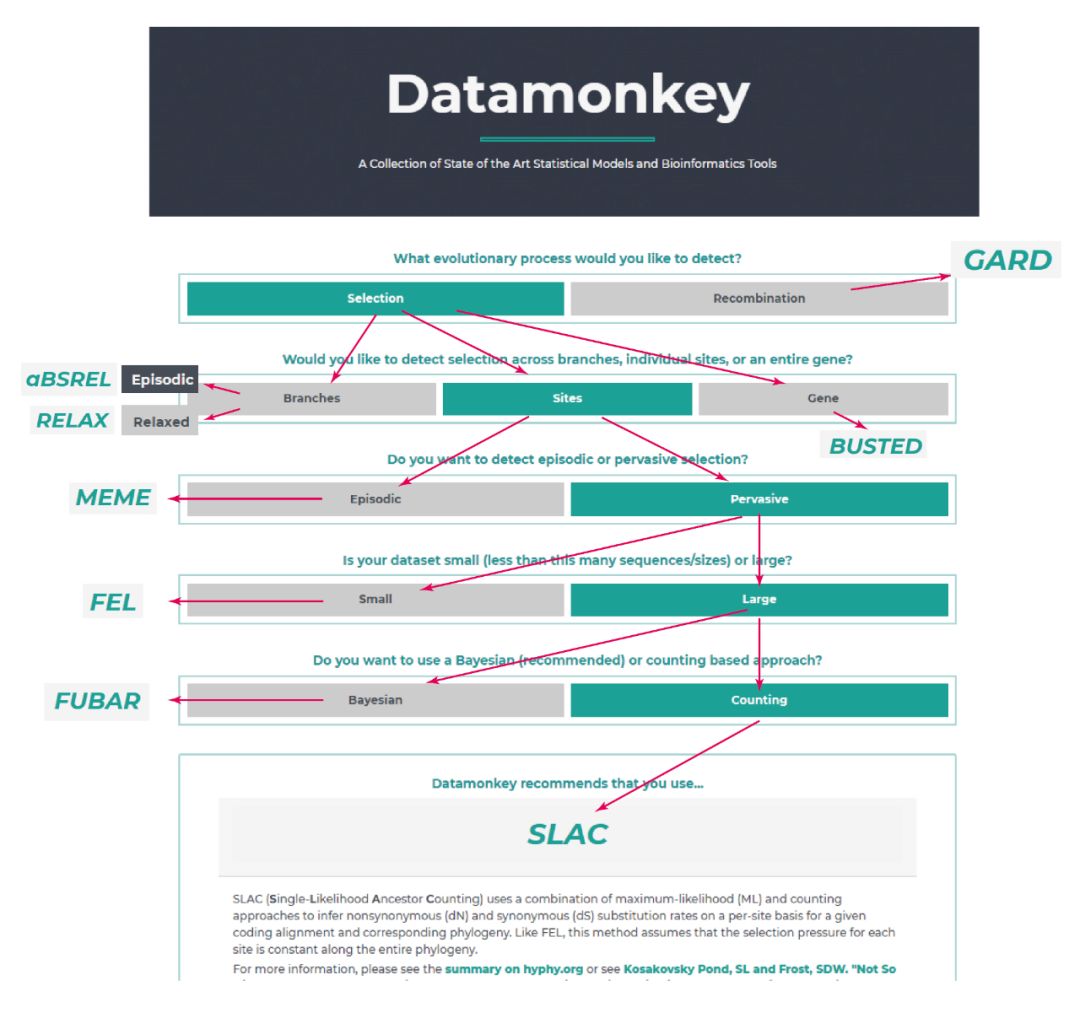
接着,用户可以选择将几种标准数据格式之一去除终止密码子的基因密码子联排文件 (NEXUS、PHYLIP、MEGA 或 FASTA) 上传。
Datamonkey 首先对上传序列进行 BLAST 搜索来聚类。使用邻接法快速重建基因家族系统发育树,并快速确定适合该基因联排的演化模型来检测基因序列中哪些位点发生了适应性进化,哪些位置与保守的功能有关。
用户可以运行不同算法如 SLAC (最多 150 条序列)、FEL (最多 50 条序列) 或 REL(最多 25 条序列) 来检测正在经历正向(适应性)进化或负向进化的位点,可以实时查询进度更新和相应结果,分析结果会在服务器上保留 96 个小时。
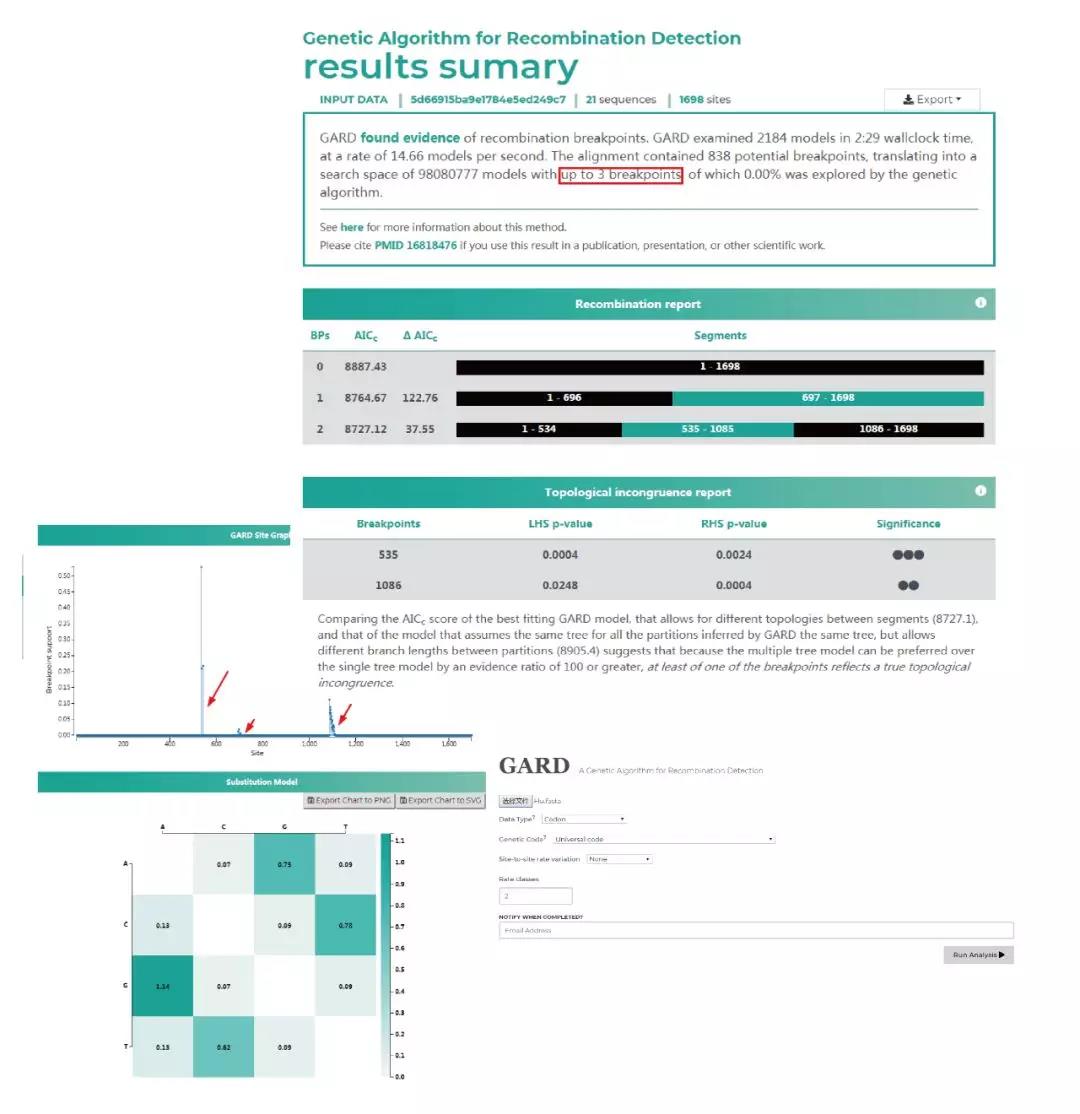
1. GARD 算法:由于发生了基因重组的序列不能用单一的系统发育史来描述,因此对这些基因的选择压力分析常常会导致假阳性。
遗传重组检测算法 GARD(Genetic Algorithm for Recombination Detection)可以分析序列中是否发生过重组,可以作为选择压力预测的预处理。如果 GARD 在数据集中检测到重组现象,它会将原始数据集分区。然后可以使用合适的分区数据集作为下一步的输入数据。
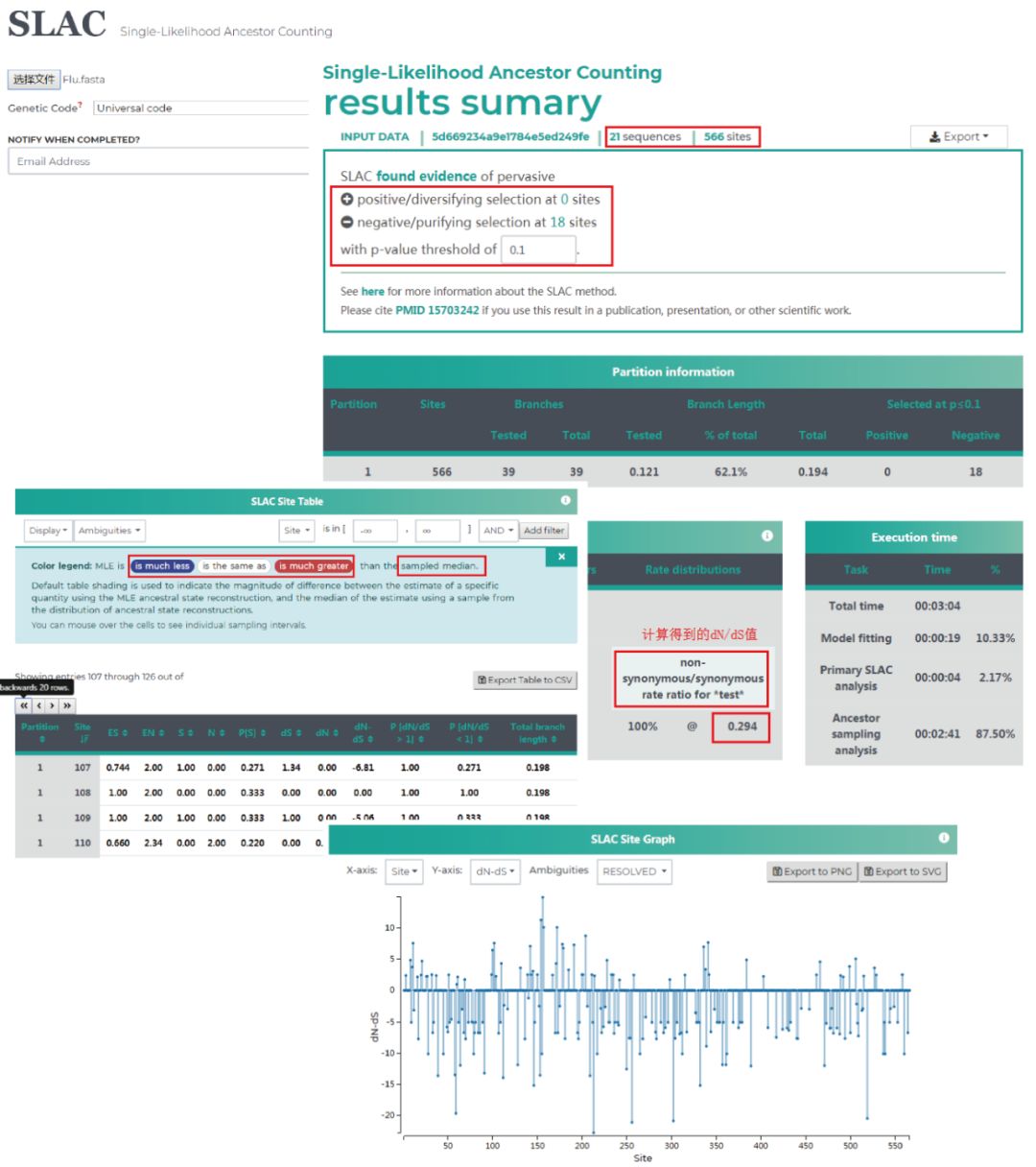
2. SLAC 方法 (Single likelihood ancestor counting) 假定每个位点的选择压力在整个系统发育过程中是恒定的。SLAC 首先在 MG94xREV 模型下优化分支长度和核苷酸替换参数,使用 ML 来推断系统发育的每个节点上最有可能的祖先序列。
然后,SLAC 直接计算每个站点上发生的非同义和同义更改的总数,在此基础上推断出非同义 (dN) 和同义 (dS) 替换率,并利用二项分布确定每个位点的显著性。
SLAC 对多序列输入文件 (>50 条) 具有良好的检测能力,可以在一分钟内完成 100 条序列和 400 个密码子的比对。对于 SLAC 的分析结果,可以沿序列查看推断发生突变的位点,也可以选择将它们映射到系统发育树上。
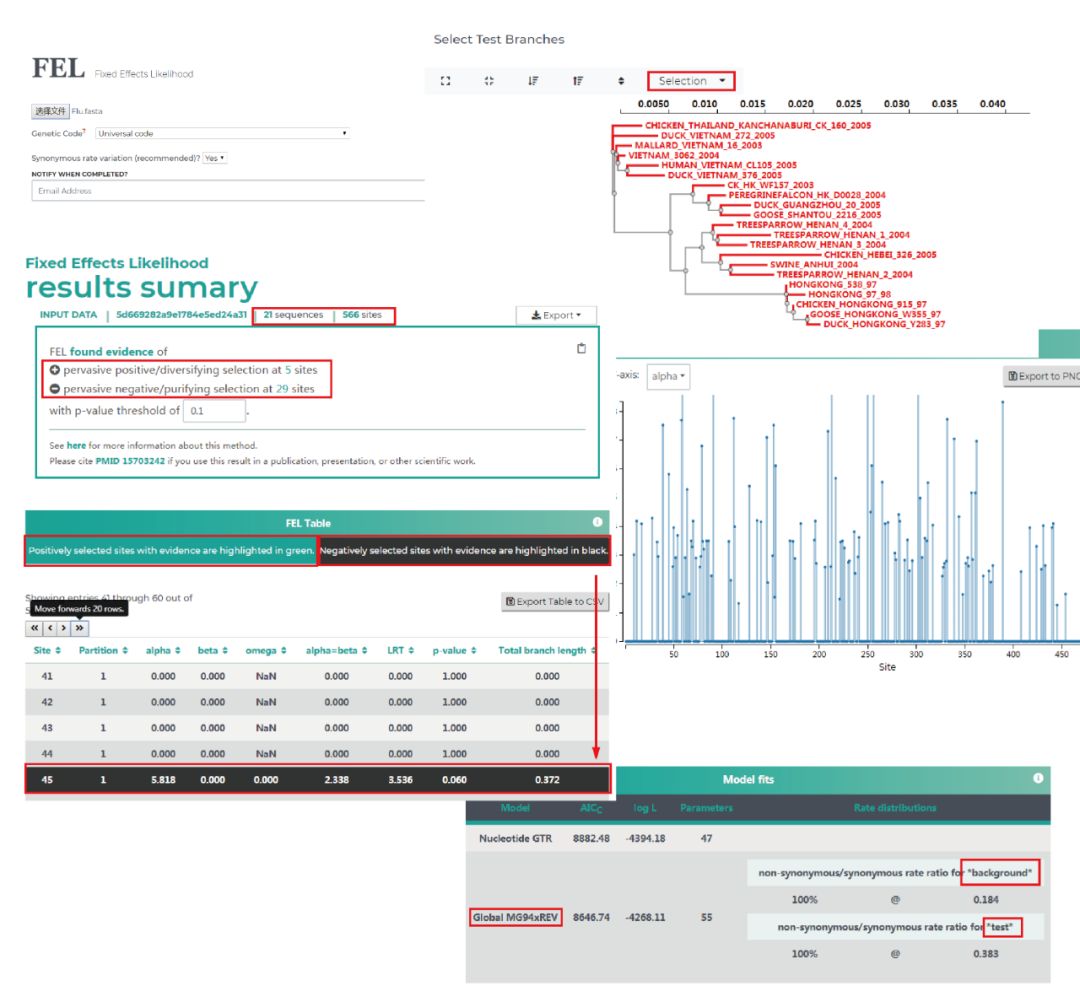
3. FEL 方法 (Fixed effects likelihood) 假设在整个系统发育过程中,每个位点的选择压力是恒定的,FEL 使用最大似然法,在优化分支长度和核苷酸替代参数后,FEL 利用 MG94xREV 模型推断每个密码子位点非同义和同义的替代率。
然后,使用似然比检验 (Likelihood Ratio Test) 对特定位点进行假设检验,以确定 dN 是否显著大于 dS。在进行此类比较时,要进行正向选择测试的谱系被标识为 foreground(前景支),与该前景谱系相比较的基因组被标记为 background(背景支)。
对于中等大小 (20 - 50 条序列) 的数据集,FEL 的鉴定结果的保守性通常比 SLAC 要低,能提供更全面的分析结果为下一步的验证提供线索。
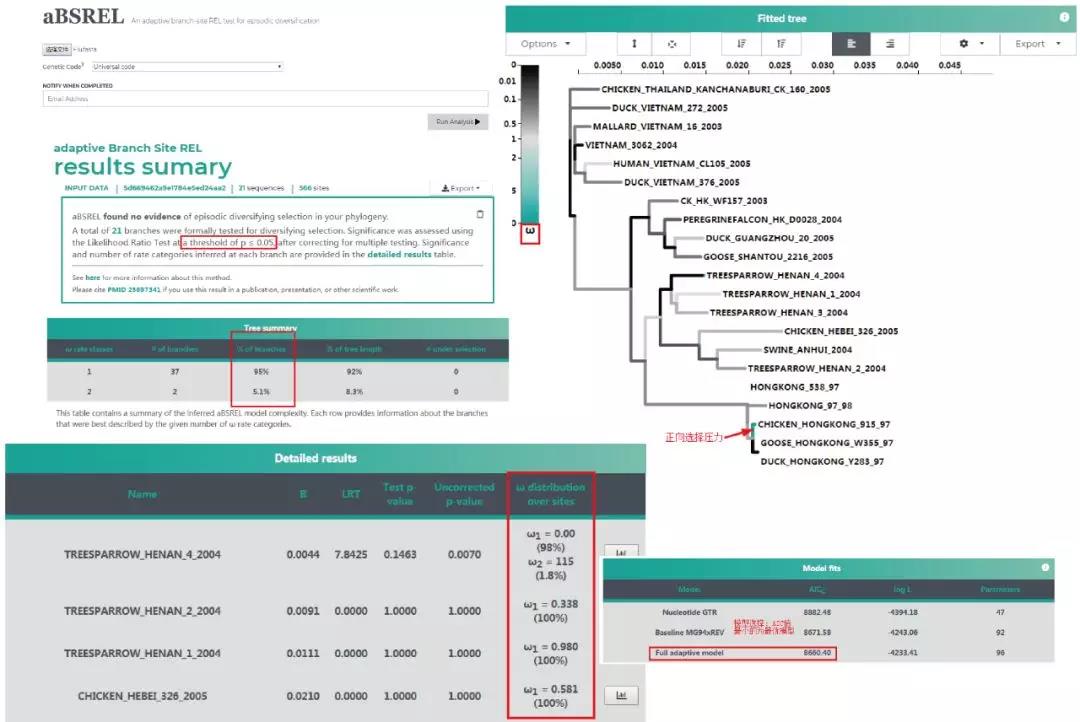
4. aBSREL (adaptive Branch-Site Random Effects Likelihood) 通过「分支-位点」模型以及似然比检验检测某些分支是否发生了正向选择。
aBSREL 预设不同的分支可能有较为复杂的进化模式,因此 aBSREL 使用 AICc 来推断每个分支 ω 的最优数量,并计算不同分支的 ω 值。
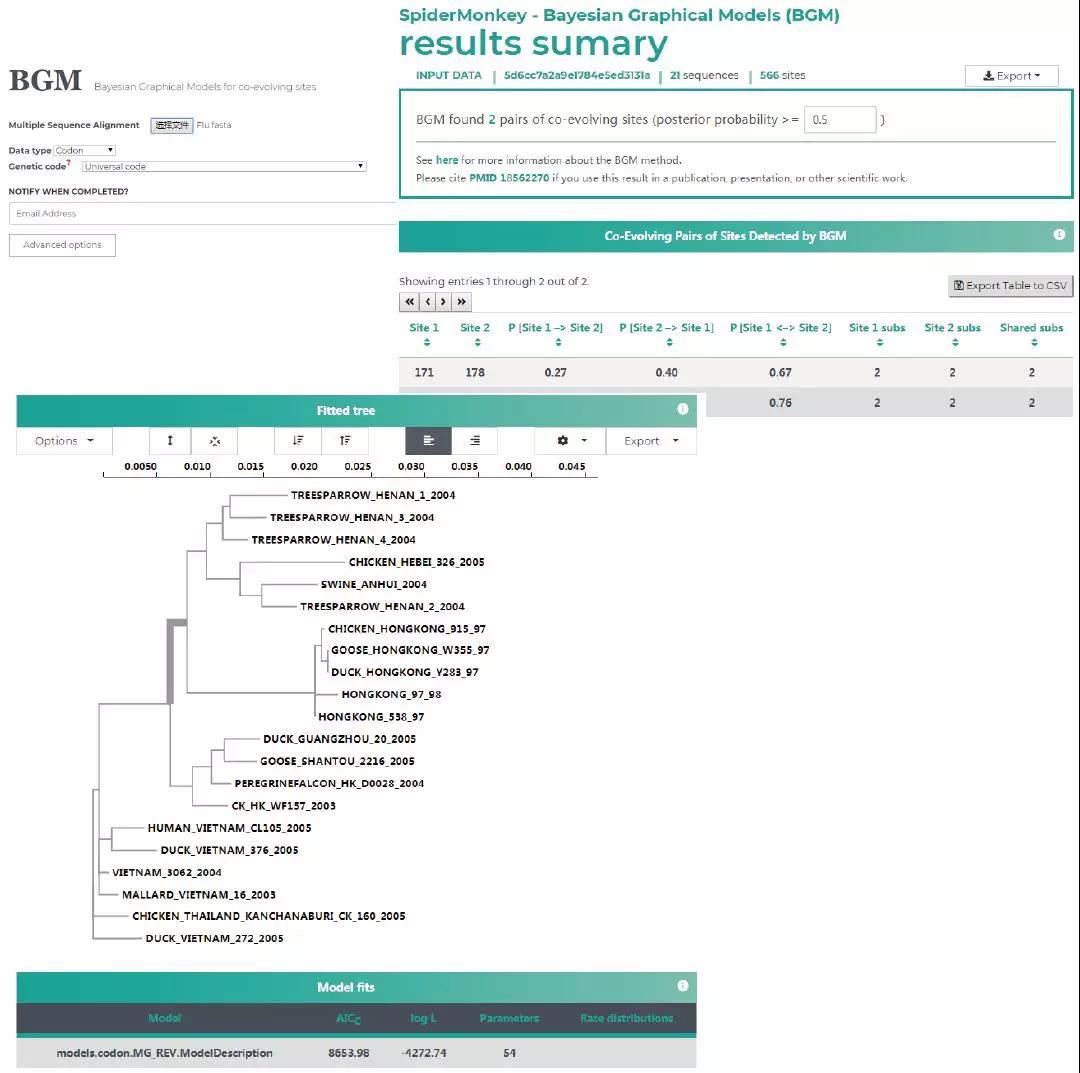
5. BGM 贝叶斯图形模型法 (Bayesian Graphical Model) 是人工智能领域的一种概率框架(probabilistic framework),它可以用来检测蛋白质中氨基酸位点间的共同进化以及相互作用关系,它会将氨基酸替换事件映射到系统发育树的分支上。

6. FUBAR (Fast, Unconstrained Bayesian AppRoximation) 使用贝叶斯法,根据给定的编码序列排列和相应的系统发育关系,推断每个位点 d 非同义替换率 (dN) 和同义替换率 (dS)。
该方法也假设在整个系统发育过程中,每个位点的选择压力是恒定的。FUBAR 使用后验概率 (posterior probabilities) 来检测结果的可靠性,一般来说后验概率>0.9 表示存在显著的正向选择。

7. FADE (FUBAR Aproach to Directional Evolution) 是在 FUBAR 引入贝叶斯框架的基础上,使用贝叶斯框架来检测蛋白质排列中的位点是否受定向选择的影响,即分析一组指定的前景支与背景支相比是否对特定的氨基酸具有替换偏好性。

8. 混合效应进化模型 MEME (Mixed Effects Model of Evolution) 采用一种混合效应极大似然方法来检验假设,即单个位点受到情景性正向选择的影响。也就是说,MEME 可以检测某些分支而不一定是全部分支受正向选择的位点。

9. RELAX 算法可以检测自然选择的强度是否在一组指定的测试分支得到了放松或增强。因此,RELAX 不是测试正向选择的合适方法。而是测试特定基因上的自然选择压力变化的趋势。
RELAX 通过引入参数 k (k ≥ 0) 来表征选择压力强度,k>1 表明测试分支的选择强度增强,而 k<1 表明,沿测试分支的选择强度减弱。

Datamonkey 主页:https://www.datamonkey.org/





