如何结合不同的g蛋白偶联受体扩展功能,并提供新的药物靶点
喝咖啡上瘾了?Rafael Franco和他的同事介绍了一种受体,这种受体是一个庞大而多样的蛋白质家族的成员——g蛋白偶联受体——是我们药店里许多药物的靶点。他们的最新发现表明,不同的腺苷受体亚型如何结合在一起,成为一种浓度感应装置,这突显出,不同的g蛋白偶联受体组合可以产生怎样的新特性,以及用药物靶向这些新特性的潜力。
1909年,剑桥生理学家约翰·纽波特·兰利(John Newport Langley)提出了一种介导药物作用的“感受性物质”。这个想法一直假设到两个进一步的突破——当雷蒙德·p·Ahlquist在1948年提出的存在两个接收器肾上腺素(α-和β-adrenoceptors),当药物能够阻止这种接收器是在1965年。阻滞剂过去是,现在仍然是对抗疾病的工具。第一个是普萘洛尔,第一临床有用的β-adrenoceptor杀杀杀,詹姆斯•布莱克在1988年被授予诺贝尔奖。
Alfred G. Gilman(1994年诺贝尔奖得主)发现G蛋白后,Langley的“接受物质”被鉴定为G蛋白偶联受体(GPCR)家族蛋白。G蛋白存在于细胞膜的内表面,并通过一种激素(如肾上腺素)与细胞表面的一个同源GPCRs结合而产生的信号传递到细胞内。
分子生物学技术已经帮助识别了氨基酸序列,结晶技术的重大进展已经帮助解决了数十个GPCRs的晶体结构,建立了它们的原型单分子结构,即一个七跨膜域蛋白与一个三聚体G蛋白偶联。2012年,诺贝尔奖授予了罗伯特·j·莱夫科维茨(Robert J. Lefkowitz)和布莱恩·k·科比尔卡(Brian K. Kobilka),表彰他们在这一蛋白质家族上的工作。
我们现在知道,GPCRs在哺乳动物基因组中高度多样化(据估计,人类大约有1000个GPCR基因——占人类蛋白质组的8% !),它们是药店中销售的约30%药物的目标。然而,尽管有了这些重要的进展,它们在介导药物作用和向细胞传递信号方面的作用仍然没有被完全了解。
作为信号开关的GPCR四重奏
我们感兴趣的是GPCRs的信号功能,尤其是对腺苷(一种最古老的激素)作出反应的受体。腺苷受体调节广泛的身体功能,包括能量和温度的稳态和神经递质释放。但有趣的是,它们还调节咖啡和可乐饮料中的咖啡因、茶中的茶碱和可可中的可可碱的作用。
与许多其他GPCRs一样,哺乳动物中的腺苷受体是相关蛋白的一个亚家族(A1、A2A、A2B和A3)。除了功能单一单位(单体)外,这些单体还可能形成由许多相等(homo)或不同(hetero)单体组成的高阶复合物(低聚体)。因此,我们考虑进行研究,以了解为什么GPCR二聚体/低聚物在自然界中存在的原因。
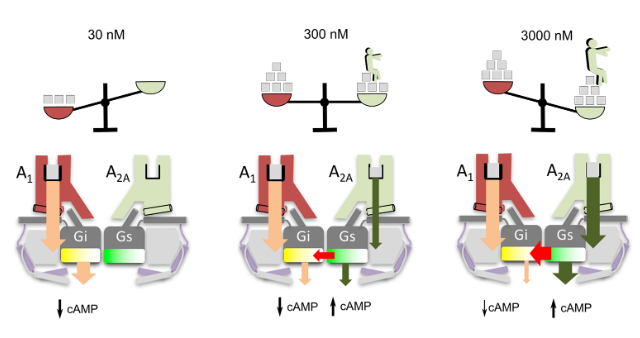
目前这一发现的起源始于12年前的一次偶然发现。为了证明A1R与Gi蛋白(介导腺苷酸环化酶的抑制和cAMP的减少)和A2AR与Gs(介导腺苷酸环化酶的激活和cAMP的增加)之间缺乏相互作用,我们发现了相反的结果。A1R和A2AR确实形成异构体,其信号输出不能作为其组分信号的总和。这赋予了A1R-A2AR复合物能够作为腺苷浓度的敏感传感器的特性,无论是在培养的细胞中还是在大脑的神经终端中。然而,实现这一目标的机制尚不清楚。
在《BMC生物学》上发表的两篇论文中,我们发现腺苷传感器的最小单元由四个受体组成,两个A1R和两个A2AR,同时与两个G蛋白(一个Gi和一个Gs)结合。结合实验和计算模型,提出了一种紧凑的菱形异质四聚体的整体结构。
在第二个贡献中,我们展示了传感器在分子水平上工作的机理。由于A1R对激素的高亲和力,低浓度的腺苷主要与A1R结合,参与gii介导的信号转导,显著减少cAMP的积累(左图)。然而,在较高的浓度下,腺苷逐渐与A2AR(中图)结合,直到饱和浓度,即A1R和A2AR都被完全占据时,由于Gs被激活而Gi被阻断,cAMP水平显著升高。我们的结构模型表明,在紧凑型四聚体中,激活的A2A受体的c末端阻止Gs和Gi的同时激活,导致优先的Gs偶联。
21世纪的一个挑战是了解其他GPCR(异型)寡聚体如何影响受体的生理作用,并评估其作为治疗靶点的潜力。高等生物中的几百个GPCRs已经提供了多样性,但我们现在还需要考虑当GPCRs形成异构体时所产生的新特性。现在已经鉴定出的异构体的数量有数百种,其中包括肽、光子和激素的受体。显然,靶向或干扰生理相关的GPCR异构体的可能性为新药物的发现提供了新的机会。