ATAC-seq利用转座酶研究染色质可接近性数据分析工具全面介绍
Assay of Transposase Accessible Chromatin sequencing(ATAC-seq)即利用转座酶研究染色质可接近性。ATAC-seq 自 2013 年开发以来广泛应用于研究有关染色质的生物学问题。根据 PubMed 上发表 ATAC-seq 有关文章可以看到,文章数量逐年上升,其中 2019 年发表文章数量最多,为 635 篇(图 1)。
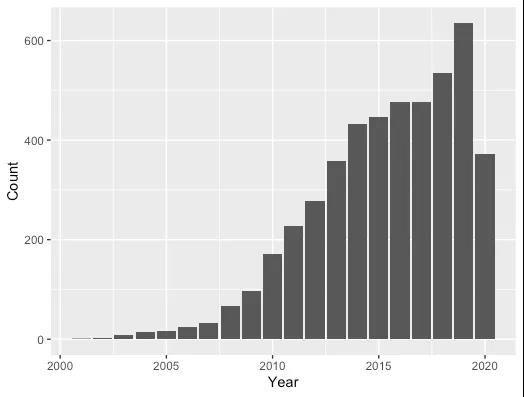
图 1
识别和表征开放染色质区域有助于确定基因组调控元件的位置和深入了解转录调控。ATAC-seq 就是 用转座酶 Tn5 获取开放染色质区域,并进行高通量测序和生物信息学分析,以此挖掘转录调控和表观修饰方面的信息。ATAC-seq 仅需要 500-50000 个细胞且操作简单有效,是当前一项重要的研究手段。但是其生物信息分析工具大多数使用 Chip-seq 和 DNas-seq 中使用的工具。而且,目前仍然没有对其分析工具的全面介绍。
2020 年 2 月 3 日,来自澳大利亚维克墨尔本莫纳什大学中央临床学院的 Nicholas C. Wong 团队在 Genome Biology 发表了 From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis 的综述性论文,主要介绍了 ATAC-seq 数据分析的主要步骤,包括预处理(质控和比对),核心分析(peak calling)和高级分析( peak 差异分析和注释,motif 分析和印迹分析),以及每一步所需的分析工具,并强调当前每一步面临的挑战,旨在为 ATAC-seq 数据分析提供一个注释指南。最后作者强调开发 ATAC-seq 特定分析工具以分析数据中的生物学意义的必要性。

图 2
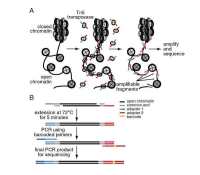
作者主要介绍 ATAC-seq 数据分析的步骤,包括预处理(质控和比对),核心分析(peak calling),peak 和 motif 相关的高级分析(差异分析,注释和 TF 印迹分析等)及其每步分析所需的软件,主要流程见图 3。
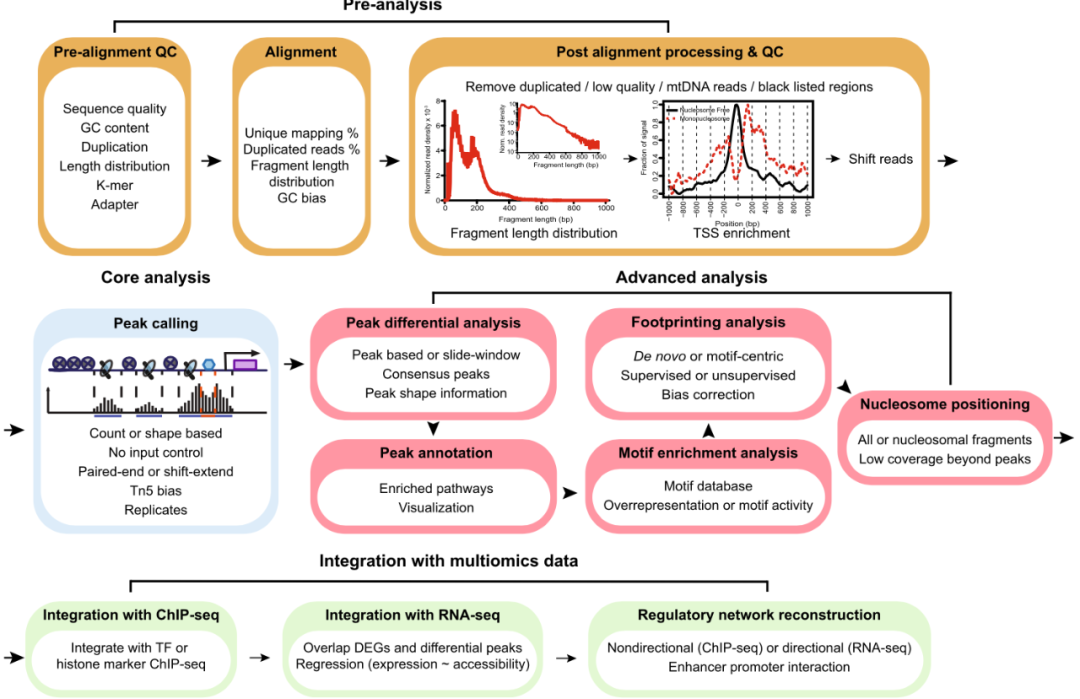
图 3 ATAC-seq 分析流程
1. 预处理:质控和比对
1.1 预处理
数据质控和比对是大部分高通量测序数据分析的标准步骤。一般使用 FastQC 对测序数据质量,GC 含量,测序数据长度分布等标准进行可视化。一般在构建文库时会添加接头序列,接头序列在数据分析时应该被去除,可使用 cutadapt,AdapterRemoval v2,Skewer 和 trimmomatic 等软件。
1.2 比对
对于短序列比对,BWA-MEM 和 Bowtie2 速度快,内存消耗少。作者建议比对效率在 80% 以上可进行下一步分析。对于哺乳动物,若进行开放染色质区域检测比对 reads 应在 50M 以上,若进行 TF 印迹分析比对 reads 应在 200M 以上。
比对后,可用 Picard 和 SAMtools 软件对比对结果进行统计,随后应去除线粒体序列和重复序列。
作者建议的预处理分析流程为:FastQC-trimmomatic-BWA-MEM-ATACseqQC。
2. 核心分析:peak calling
ATAC-seq 数据分析的第二个主要步骤是鉴定染色质可接近区域(peak calling)。当前,MACS2 是 peak caller 的默认软件,是唯一一个 ATAC-seq 数据分析的特有软件,其他软件也都适用于 ChIP-seq 和 DNase-seq。peak caller 软件可以分为 count-based,shape-based 和 Markov model 三种类型(图 4a)。
3. 高级分析
3.1 peak 差异分析
目前,还没有专门为 ATAC-seq 数据分析开发的 peak 差异分析工具。一种直接的方法是找到候选区域进行归一化,并计算这些区域的片段并与其他情况进行统计比较。这些操作都可以手动完成也可以使用自动化工具来实现(图 4b)。
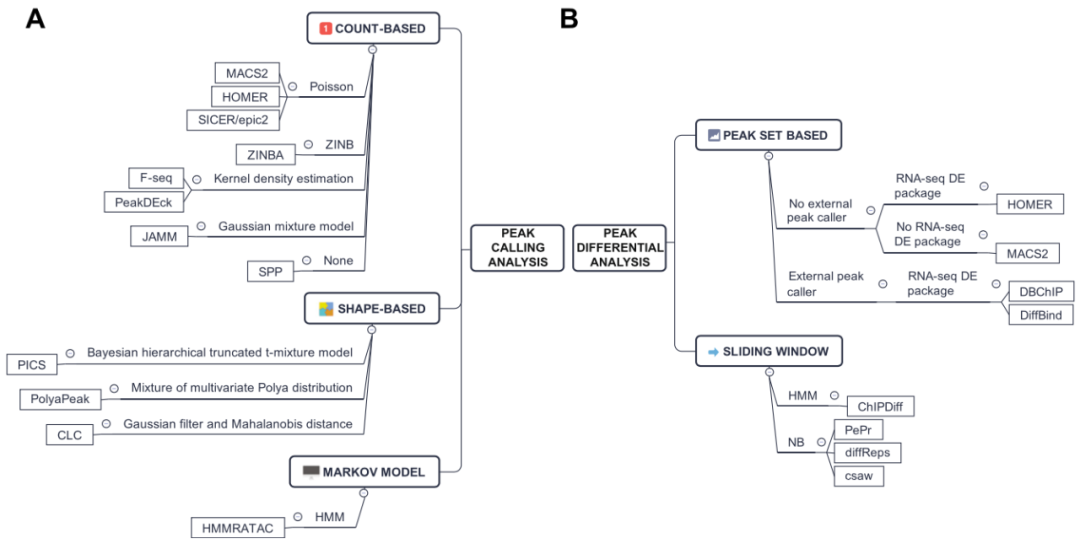
图 4 peak calling 和 peak 差异分析工具总结
3.2 peak 注释
在获得 peak 集后,可通过对 peak 注释来了解染色质可及性和基因调控的关系。一般来说,是将 peak 注释到距离最近的基因或调控元件。常用软件有 HOMER,ChIPseeker 和 ChIPpeakAnno,其中 ChIPseeker 和 ChIPpeakAnno 可对注释结果进行可视化。GO,KEGG 注释可对 peak 集进行功能富集分析。一般来说 peak 注释有助于了解他的生物学功能特点。
3.3 motif
尽管 peak 注释可以提供功能信息但是它仍然不能解释潜在的调控机制。开放染色质通过 TF 影响转录调控。TF 结合的 DNA 序列称为 motif,结合位置称为 TF 结合位点(TFBS)。因此了解 motif 作用和活性的改变有助于阐明潜在的调控网络也可以鉴定关键的调控因子。基于 motif 或 TF 分析方法有两种:基于序列的 motif 频率或活性预测和 TF 印迹分析。
为了充分探索 motif 信息,研究人员从实验方法和计算方法两方面着手建立了 motif 数据库。最有名的 motif 数据库是 JASPAR,包含多个物种。Motif 信息主要以文本格式存储,例如位置权重矩阵(PWM)。HOMER 和 R 包 TFBSTools 及 motif-matchr 工具可以使用 PWM 搜索给定的 DNA 序列来推断 TFBSs。PWMScan 和 MEME suite 也广泛应用于 motif 搜索。
3.4 TF 印迹分析
另一个阐明 TF 调控的方法是 TF 印迹法。ATAC-seq 中的印迹是指一种模式,该模式是指激活 TF 结合 DNA 序列并在结合位点阻止 Tn5 切割。因此,TFs 印迹分析可以重构特定样本的调控网络。TF 印迹分析工具主要分为两类:de novo 和 motif-centric。de novo 可根据典型的印迹模式(peak-dip-peak)预测所有的印迹。随后这些推断的印迹位点匹配已知的 motif 或识别新的 motif。相反的 motif-centric 方法需要预先输入 TFBSs 并使用监督或非监督方法区分这些位点是否与 DNA 序列结合(表 1)。
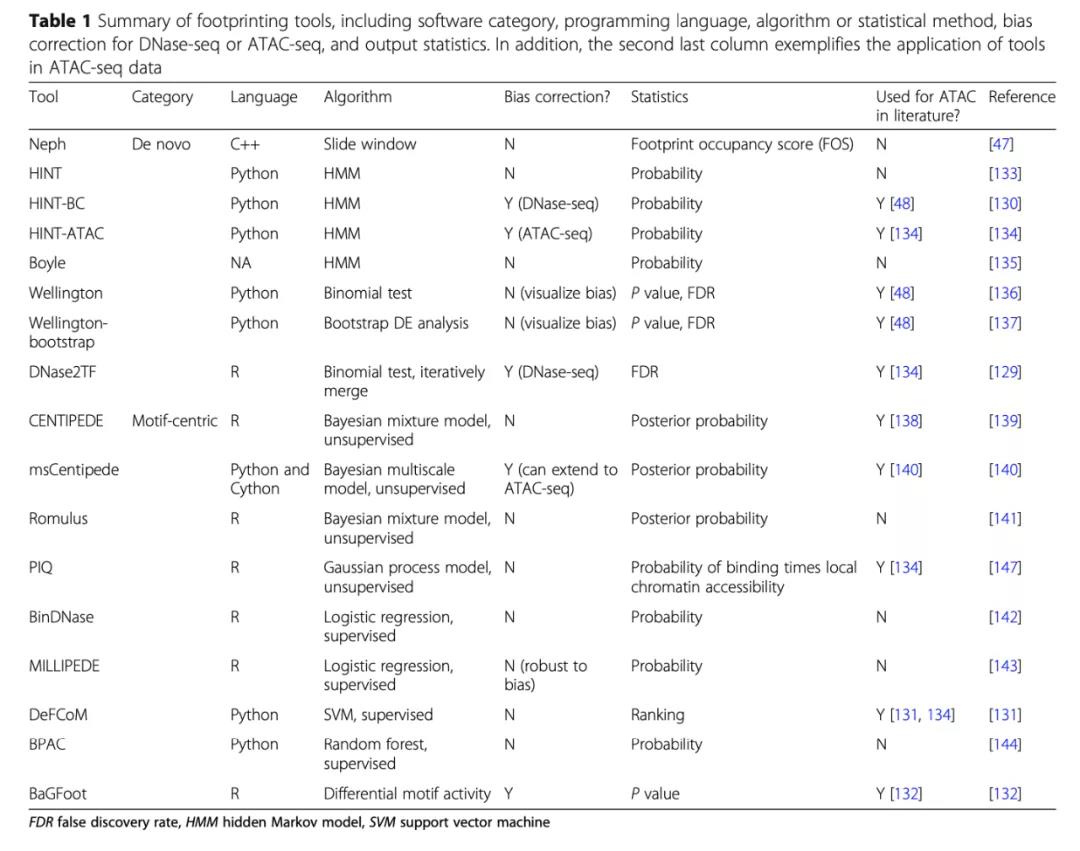
表 1 TF 印迹工具总结
总结
随着处理 ATAC-seq 的数据分析流程的需求逐渐增加,目前已有一些针对不同下游分析的流程开发出来。esATAC 和 CIPHER 重点关注 peak 注释而 GUAVA 重点关注差异 peak 鉴定和功能注释,ATAC2GRN 重点优化了 TF 印迹分析部分。这些分析流程为没有编程能力的研究人员分析 ATAC-seq 数据提供了方便。但是这些分析流程的灵活性较差,不能随意的调整参数,大多数参数都是根据经验硬性定义的。总的来说,这些具有可视化界面流程更适合非程序员使用。
ATAC-seq 近几年发展迅速并成为了研究染色质可及性的有效方法。本文,作则系统的讨论了 ATAC-seq 分析的所有主要步骤并提供了每步所需工具的指南。ATAC-seq 作为一种信息丰富的分析方法,在分析染色质状态,TF 印迹和调控网络重建方面需要大量的生物信息学分析工具。在此基础上,作者建议研究人员结合 FastQC、trimmomatic 和 BWA-MEM 进行预分析,MACS2 进行 peak calling。
对于高级分析,作者建议使用 csaw 进行 peak 差异分析 MEME situe 进行 motif 检测和富集分析,ChIPseeker 进行注释和可视化,HINT-ATAC 进行 TF 印迹分析。但是,对于每一步,研究人员都可参考本文来选择替代工具,作者建议根据实验背景和所收集的数据来选择工具。