合作专家 | 吴宇晨硕士
生物学 厦门大学
审核专家 | 王淼博士
病原微生物 中国农业科学院
简介
利用一次 PCR 方法进行定点突变分子克隆,较盒式突变及其它传统方法更快速简便。
原理
PCR 原理: 在 DNA 聚合酶催化下,以母链 DNA 为模板,以特定引物为延伸起点,通过变性、退火、延伸等步骤,体外复制出与母链模板 DNA 互补的子链 DNA 的过程。
DPNⅠ酶消化原理:DPN1 是一种限制性核酸内切酶,能够特异性切除甲基化的 DNA 链。定点突变后,我们需要区分并去除原始 DNA(未突变模板)。模版质粒来源于大肠杆菌,是经 dam 甲基化修饰的, DpnⅠ识别甲基化的 GATC 中的 A,而 GATC 在几乎各种质粒中都会出现,且不止一次,因而模板链对 DpnI 敏感而被切碎;而体外合成的带突变序列的质粒来源于 PCR 没有被甲基化,不能被 DpnⅠ识别剪切,因此在随后的转化中得以成功转化,即可得到突变质粒的克隆。
用途
用于在质粒中引入少数碱基的定点突变。
材料与仪器
步骤
一、设计引物
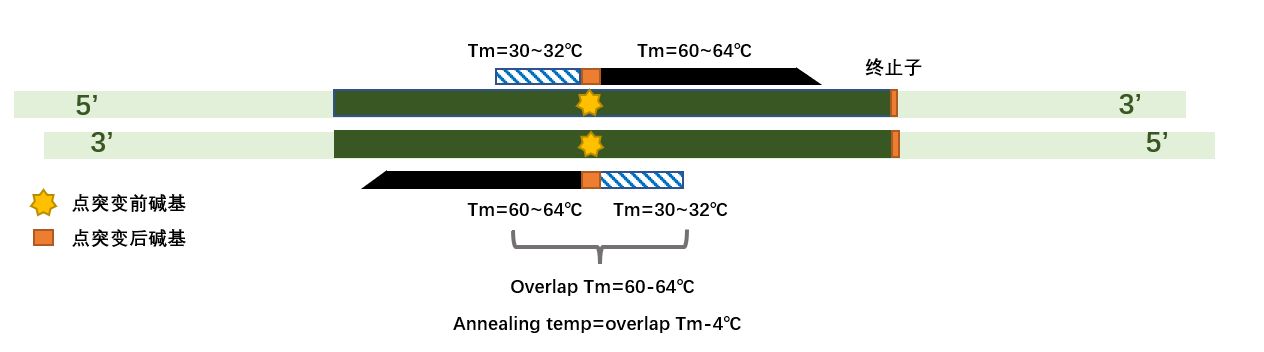
如图在欲突变位点设计 PCR 定点突变所需引物,设计的引物需含有突变后碱基序列,订购引物。
二、质粒 PCR
将稀释好的质粒模板与订购的引物以及其他试剂按如下比例混合(50 μl):
|
10 μM 正向引物 |
1 μl |
|
10 μM 反向引物 |
1 μl |
|
模板 cDNA(20~200 μM) |
2 μl |
|
2×高保真PCR混合酶 |
25 μl |
|
去离子水 |
21 μl |
将混合好的体系进行 PCR,程序如下:
|
① 预变性 |
94 ℃ |
1 min |
|
② 变性 |
98 ℃ |
10 s |
|
③ 退火 |
58 ℃ |
30 s |
|
④ 延伸 |
72 ℃ |
X min |
|
⑤ 充分延伸 |
72 ℃ |
10 min |
|
⑥ 保存 |
4 ℃ |
∞ |
步骤 ②-④ 重复 28 次循环。 根据酶的扩增效率及目的片段大小选择延伸时间。例如:酶的扩增效率为 1 000 bp/min,质粒大小为 9 000 bp,则 X 为 9 min。
三、DPNⅠ 酶消化
将 PCR 后的产物和 DPNⅠ 按一下比例混合,消化模板质粒:
|
PCR 产物 |
4 μl |
|
DPNⅠ |
0.5 μl |
|
cut buffer |
1 μl |
|
去离子水 |
4.5 μl |
四、转化
将连接好的完整质粒,转化 DH5-α 感受态细胞中,冰上孵育 30 min,42 ℃ 热击 90 s,再置于冰上冷却 5 min,完成转化。用稀释涂布法将感受态溶液涂至含有相应抗性的 LB 固体培养基表面, 37 ℃ 培养箱中过夜培养,次日会长出菌落,挑取菌落进行测序。
注意事项
1、模板投放量不应过高,会使得消化不完全,长出未突变的菌株。
2、循环数不应该太多,18 个循环足够,由于 PCR 区域是整个质粒,时长会很久,在循环后期酶的活性可能会下降,保真性变差。
常见问题
1、PCR 后无条带,可能是引物设计中 GC 含量过高,导致引物碱基数过少,应尽量延长引物长度。
2、若测序阳性率低,检查模板投放量是否过多,或 DPNⅠ 酶失活,使得消化不完全,长出未突变的菌株。
3、若未长出菌落,检查模板质粒是否变质,或实验过程是否有其他意外。
来源:丁香实验







