简介
分子标记(molecular marker)是遗传标记(genetic marker)的一种,是在基因水平上的标记,直接在 DNA 分子上检测遗传变异,用作指示基因组范围变化的多态性标记。分子标记能对不同发育时期的个体、任何组织器官甚至细胞作检测,数量极多,遍及整个基因组,多态性高,遗传稳定,不受环境及基因表达与否的限制,正是因为这些优点,分子标记的应用越来越广泛。它包括 RFLP、RAPD、AFLP、SSR 等。
长期以来,植物学研究中选择都是基于表型性状、形态学、结构等方面进行的,当性状的遗传基础较为简单或即使较为复杂但表现加性基因遗传效应时,表型选择是有效的。但物种的许多重要表型性状为数量性状,如产量等;或多基因控制的质量性状,如抗性等;或表型难以准确鉴定的性状,如根系活力等。此时根据表型提供的对性状遗传潜力的度量是不确切的,因而选择是低效的。分子生物学技术的发展为植物科学研究提供了一种基于 DNA 变异的新型遗传标记——DNA 分子标记,或简称分子标记。与传统应用的常规遗传标记相比,分子标记具有许多明显的优点,因而已被广泛应用于现代植物研究的各个方面,大量以前无法进行的研究目前利用分子标记手段正蓬勃开展,并取得了丰硕的成果。尤其是当分子标记技术与传统形态结构紧密结合后,正在为植物科学技术带来一场新的变革。
分子标记大多以电泳谱带的形式表现,大致可分为 3 大类:第一类是以分子杂交为核心的分子标记技术,包括限制性片段长度多态性(restriction fragment length polymorphism,RFLP)标记、DNA 指纹(DNA fingerprinting)技术、原位杂交(in situ hybridization)等;第二类是以聚合酶链反应(polymerase chain reaction,PCR)反应为核心的分子标记技术,包括随机扩增多态性 DNA(random amplification polymorphism DNA,RAPD)标记、简单序列重复(simple sequence repeat,SSR)标记或简单序列长度多态性(simple sequence length polymorphism,SSLP)标记、扩增片段长度多态性(amplified fragment length polymorphism,AFLP)标记、序标位(sequence tagged sites,STS)标记、序列特征化扩增区域(sequence charactered amplified region,SCAR)标记等;第三类是一些新型的分子标记,如单核昔酸多态性(single nucleotide polymorphism,SNP)标记、表达序列标签(expressed sequences tags,EST)标记等。以下将介绍其中最常用的一些标记技术。
原理
随机扩增多态 DNA 标记由 Williams 等于 1990 年创立。其基本原理与 PCR 技术一致。PCR 技术是一种体外快速扩增特异基因或 DNA 序列的方法,由 Mullis 等于 1985 年首创。该技术在试管中建立反应体系,经数小时后,就能将极微量的目的基因或某一特定的 DNA 片段扩增数百万倍。其原理与细胞内发生的 DNA 复制过程相类似,首先是双链 DNA 分子在邻近沸点的温度下加热时分离成两条单链 DNA 分子,然后 DNA 聚合酶以单链 DNA 为模板,并利用反应混合物中的 4 种脱氧核昔三磷酸(dNTP)合成新生的 DNA 互补链,以上过程为一个循环,每一个循环的产物可以作为下一个循环的模板,经过 20~30 个循环后,介于两个引物间的特异 DNA 片段以几何数级得以大量复制。RAPD 标记技术就是用一个(有时用两个)随机引物(一般 8~10 个碱基)非定点地扩增基因组 DNA,然后用凝胶电泳分开扩增片段。遗传材料的基因组 DNA 如果在特定引物结合区域发生 DNA 片段插入、缺失或碱基突变,就有可能导致引物结合位点的分布发生相应的变化,导致 PCR 产物增加、缺少或发生分子质量变化。若 PCR 产物增加或缺少,则产生 RAPD 标记。随机扩增多态性 DNA 分子标记,简称 RAPD 标记,常常呈共显性遗传。应用 RAPD 技术进行品种纯度鉴定,方法简单、快捷、可靠,不需要任何前期 DNA 模板信息。对于任一特定的 RAPD 引物,即随机引物在模板的两条链上有互补的位置,且引物的 3,端相距在一定的长度范围之内,就可以扩增出来 DNA 片段。通过对 PCR 产物的检测分析即可以测出基因组在这些区域的多态性(图 42-5)。
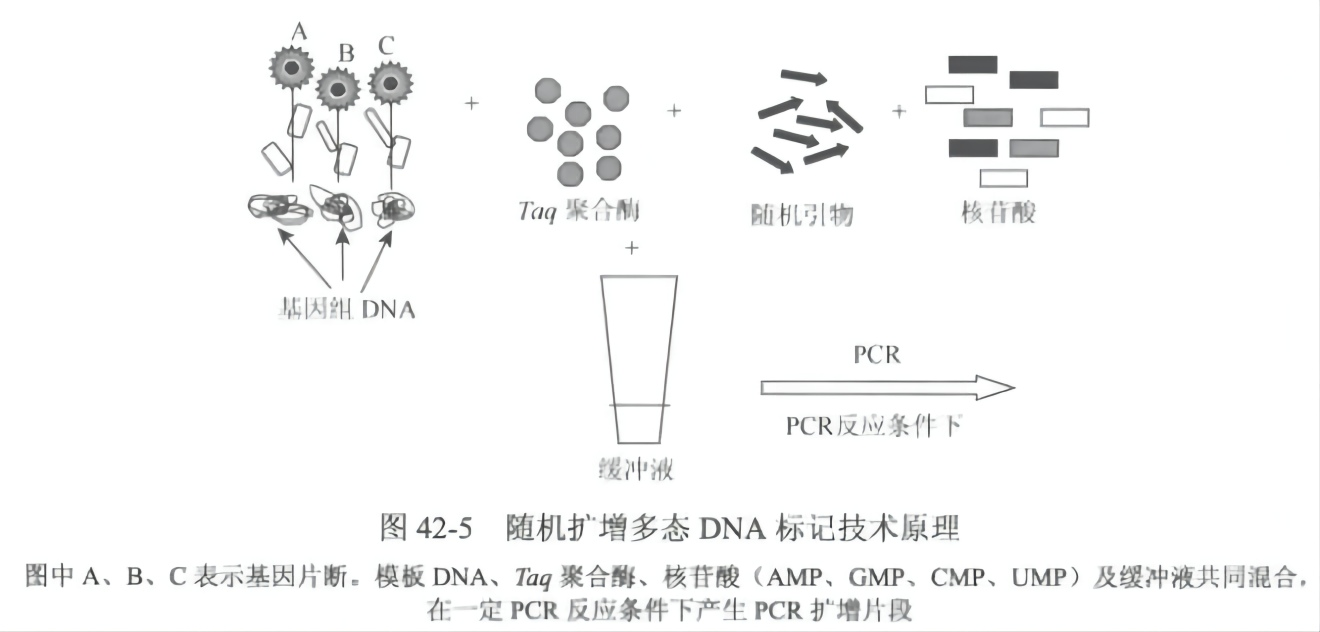
RAPD 标记的主要特点:① 不需 DNA 探针,设计引物也无需知道序列信息。② 技术简便,检测速度快;不涉及分子杂交和放射性自显影等技术。③ DNA 样品需要量少,引物价格便宜,成本较低。④ 不依赖于种属特异性和基因组结构,一套引物可用于不同生物基因组分析。
RAPD 标记缺点:① RAPD 标记是一个显性标记,不能鉴别杂合子和纯合子。②存在共迁移问题,凝胶电泳只能分开不同长度 DNA 片段,而不能分开那些长度相同但碱基序列组成不同的 DNA 片段。③ RAPD 标记技术中影响因素很多,因此实验的稳定性和重复性差,结果可靠性较低。
材料与仪器
步骤
(1)选择16株植物新鲜叶片样品,通过40条随机引物的RAPD-PCR实验,筛选出理想的随机引物(表42-1)。
|
名称 |
序列5'一3' |
名称 |
序列5'一3' |
名称 |
序列5'一3' |
名称 |
序列5'一3' |
|
A01 |
CACK1CCCTTC |
A11 |
CAATCGCCGT |
D01 |
ACCGCGAAGG |
D11 |
AGCGCCATTG |
|
A02 |
TGCCGAGCTG |
A12 |
TCGGCGATAG |
D02 |
GGACCCAACC |
D12 |
CACCGTATCC |
|
A03 |
AGTCAGCCAC |
A13 |
CAGCACCCAC |
D03 |
GTCGCCGTCA |
D13 |
GGGGTGACGA |
|
A04 |
AATCGGGCTG |
A14 |
TCTGTGCTGG |
D04 |
TCTGGTGAGG |
D14 |
CTTCCCCAAG |
|
A05 |
AGGGGTCTTG |
A15 |
TTCCGAACCC |
D05 |
TGAGCGGACA |
D15 |
CATCCGTGCT |
|
A06 |
GGTCCCTGAC |
A16 |
AGCCAGCGAA |
D06 |
ACCTGAACGG |
D16 |
AGGGCGTAAG |
|
A07 |
GAAACGGGTG |
A17 |
GACCGCTTGT |
D07 |
TTGGCACGGG |
D17 |
TTTCCCACGG |
|
A08 |
GTGACGTAGG |
A18 |
AGGTGACCGT |
D08 |
GTGTGCCCCA |
D18 |
GAGAGCCAAC |
|
A09 |
GGGTAACGCC |
A19 |
CAAACGTCGG |
D09 |
CTCTGGAGAC |
D19 |
CTGGGGACTT |
|
A10 |
GTGATCGCAG |
A20 |
GTTGCGATCC |
D10 |
GGTCTACACC |
D20 |
ACCCGGTCAC |
资料来源:赵鹏等,2012b。
(2)对 16 株叶片进行基因组 DNA 提取。
(3)在 20 μL 的反应体系中加入以下物质:模板DNA 2μL,随机引物1 μmol/L,10 × PCR Buffer 2.0 μL,MgCl2 2.5 mmol/L,dNTP(dATP、dCTP、dGTP、dTTP)各 0.25mmol/L,Taq 聚合酶 0.5 U。混匀稍离心(引物见表42-1)o
(4)在加热至 90 ℃ 以上的 PCR 仪中 95 ℃ 预变性 3 min,然后循环:94 ℃,40 s;36 ℃,1 min,72 ℃,2 min,共 30 个循环。
(5)循环结束后,72 ℃,10 min进行延伸,4 ℃保存。
(6)取PCR产物 5 μL 加 1 μL 上样缓冲液(6×)于 2% 琼脂糖凝胶上电泳,稳压 100 V。
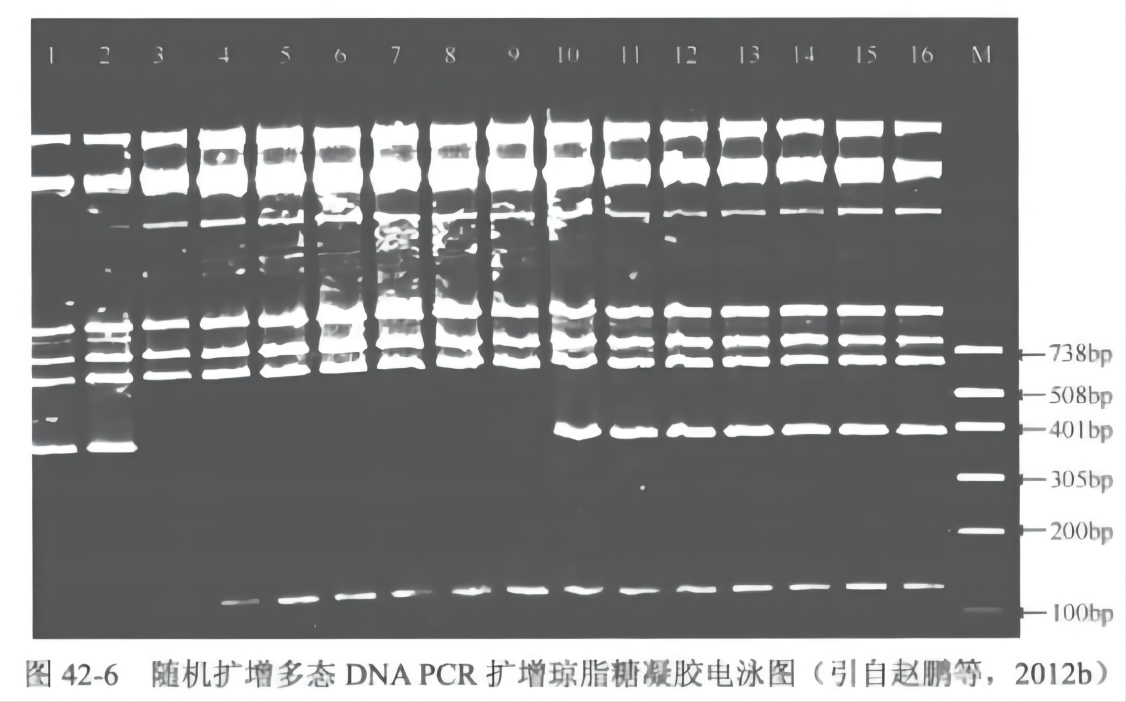
(7)电泳结束,观察、拍照(图 42-6)。
(8)根据 RAPD 扩增结果计算:遗传相似性系数 S = 2Nxy/(Nx + Ny),Nxy 为种间共有的扩增带,Nx 为 X 种具有的扩增带,Ny 为 Y 种具有的扩增带;遗传距离(D):D = 1 - S。
来源:丁香实验