简介
分子标记(molecular marker)是遗传标记(genetic marker)的一种,是在基因水平上的标记,直接在 DNA 分子上检测遗传变异,用作指示基因组范围变化的多态性标记。分子标记能对不同发育时期的个体、任何组织器官甚至细胞作检测,数量极多,遍及整个基因组,多态性高,遗传稳定,不受环境及基因表达与否的限制,正是因为这些优点,分子标记的应用越来越广泛。它包括 RFLP、RAPD、AFLP、SSR 等。
长期以来,植物学研究中选择都是基于表型性状、形态学、结构等方面进行的,当性状的遗传基础较为简单或即使较为复杂但表现加性基因遗传效应时,表型选择是有效的。但物种的许多重要表型性状为数量性状,如产量等;或多基因控制的质量性状,如抗性等;或表型难以准确鉴定的性状,如根系活力等。此时根据表型提供的对性状遗传潜力的度量是不确切的,因而选择是低效的。分子生物学技术的发展为植物科学研究提供了一种基于 DNA 变异的新型遗传标记——DNA 分子标记,或简称分子标记。与传统应用的常规遗传标记相比,分子标记具有许多明显的优点,因而已被广泛应用于现代植物研究的各个方面,大量以前无法进行的研究目前利用分子标记手段正蓬勃开展,并取得了丰硕的成果。尤其是当分子标记技术与传统形态结构紧密结合后,正在为植物科学技术带来一场新的变革。
分子标记大多以电泳谱带的形式表现,大致可分为 3 大类:第一类是以分子杂交为核心的分子标记技术,包括限制性片段长度多态性(restriction fragment length polymorphism,RFLP)标记、DNA 指纹(DNA fingerprinting)技术、原位杂交(in situ hybridization)等;第二类是以聚合酶链反应(polymerase chain reaction,PCR)反应为核心的分子标记技术,包括随机扩增多态性 DNA(random amplification polymorphism DNA,RAPD)标记、简单序列重复(simple sequence repeat,SSR)标记或简单序列长度多态性(simple sequence length polymorphism,SSLP)标记、扩增片段长度多态性(amplified fragment length polymorphism,AFLP)标记、序标位(sequence tagged sites,STS)标记、序列特征化扩增区域(sequence charactered amplified region,SCAR)标记等;第三类是一些新型的分子标记,如单核昔酸多态性(single nucleotide polymorphism,SNP)标记、表达序列标签(expressed sequences tags,EST)标记等。以下将介绍其中最常用的一些标记技术。
原理
限制性片段长度多态性标记是以分子杂交为核心的分子标记技术,该技术由 Grodzicker 等于 1974 年创立。特定生物类型的基因组 DNA 经某一种限制性内切酶完全酶解后,会产生分子质量不同的同源等位片段,或称限制性等位片段。RFLP 标记技术的基本原理就是通过电泳的方法分离和检测这些片段。凡是可以引起酶解位点变异的突变,如点突变(新产生和去除酶切位点)和一段 DNA 的重组(如插入和缺失造成酶切位点间的长度发生变化)等均可导致限制性等位片段的变化,从而产生 RFLP(图 42-3)。该分子标记是不依赖于 PCR 扩增的一种分子生物学研究技术。RFLP 分子标记不仅具有稳定遗传、高度共显性和多态性等特性,而且还能对不同的植物类群进行直接的差异分析比较(图 42-4)。

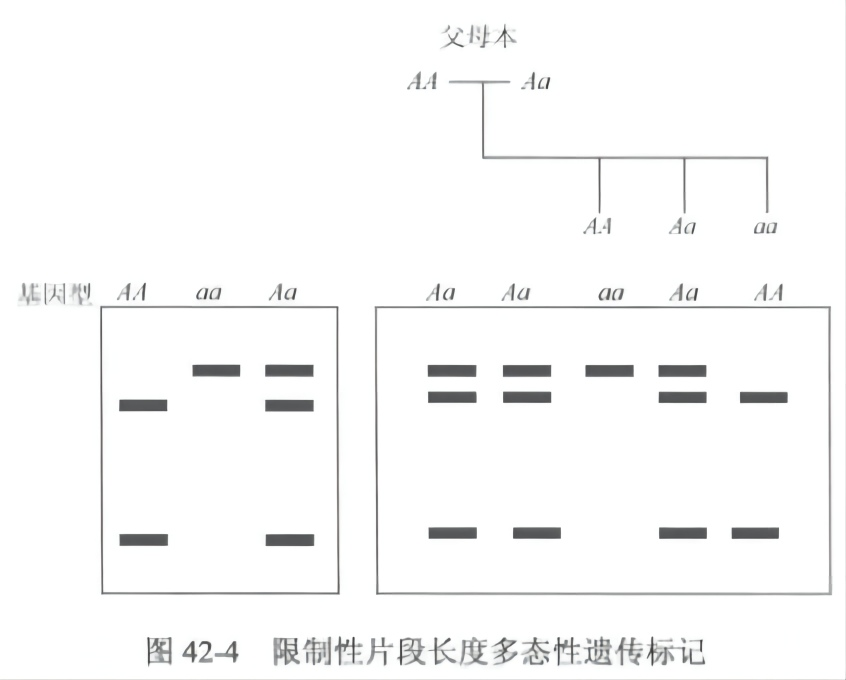
RFLP 标记的主要特点:① 遍布于整个基因组,数量几乎是无限的;② 无表型效应,不受发育阶段及器官特异性限制;③ 共显性,可区分纯合子和杂合子;④ 结果稳定、可靠;⑤ DNA 需要量大,检测技术繁杂,难以用于大规模的育种实践。
RFLP 标记的优点:① 标记数目可以是无限的。RFLP 揭示的是 DNA 水平自然变异,其数目几乎是无限的。② 大部分标记为共显性:表现 RFLP 的位点一般是单一序列,每个位点通常有两个等位基因(其显性)。遵循孟德尔式遗传,因而 RFLP 标记图也可用传统的遗传图谱方法来构建。③ 任何发育期都可预测,不受环境影响。DNA 分子水平标记没有发育阶段或器官的特异性,不受环境条件及基因互作的影响。④ 高度变异性。每一植株都会有大量的多态性。通常只要有一次有性杂交,一个作图群体就能构建一个较丰富的 RFLP 图谱。
在做 RFLP 分析技术时,有几个问题需要引起密切注意:① 作为检测对象的 DNA 分子必须保持大分子,在抽提 DNA 的过程中避免人为地将 DNA 分子机械性切割成小片段,否则最终显示的 RFLP 图谱可能是一种假象。② 在用限制性内切酶消化大分子 DNA 时,要使 DNA 被完全消化,否则所得的结果也不可靠。③ 被消化的 DNA 浓度不能太高。④ 电泳时要用低压电泳。⑤ 杂交前探针必须充分变性。⑥ 要根据探针标记的情况,以及探针与靶 DNA 间序列互补的程度和 G、C 的含量来掌握杂交和洗膜的条件。⑦ 作放射自显影时,要根据杂交后膜上的放射活性等因素决定曝光的时间。
材料与仪器
步骤
限制性片段长度多态性标记的基本过程可分为如下几步:
(1)DNA 提取(靶 DNA 的准备):先将基因组 DNA 抽提出来,选用合适的限制性内切核酸酶酶解基因组 DNA,将酶解出来的具有各种长度的 DNA 片段在琼脂糖凝胶电泳分离,使其按片段的长短排列,将 DNA 片段变性后转移至硝酸纤维素膜或尼龙膜上,称印迹(Southern blot)转移,并在 80 ℃ 烘烤或用长波紫外线照射,将 DNA 固定在膜上。
(2)核酸探针的标记:将准备作为探针的 DNA 片段纯化(这些 DNA 片段可以是基因组 DNA 的一个片段,或是 cDNA,或是人工合成的寡核苷酸),用放射性元素(如 a-32P)或非放射性元素(如 Dig-dUTP 等)标记,经纯化后再用。探针的获得是最先用内切酶处理植物的 DNA 获得 DNA 片段,再将其重组到质粒上,使它能插入细菌寄主细胞并在里面进行复制,通过稀释繁殖,每个菌落一般由携带某一段 DNA 的细菌繁殖而来,这种在细菌细胞中扩增、纯化 DNA 片段的过程称为 DNA 克隆。这一系列的克隆经放射性同位素标记就成了一系列的探针。
(3)杂交显示:将标记好的探针与硝酸纤维素膜或尼龙膜上的单链核酸杂交,洗膜去除未杂交的标记探针后,进行放射自显影或加入酶的底物进行显色反应,再对显示出来的谱带进行分析。
来源:丁香实验