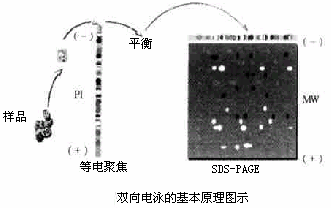
简介
高分辨的双向聚丙烯酰胺凝胶电泳(2D-PAGE)首先由 O'Farrell 于 1975 年创立,后经过 Anderson 实验室及其他实验室的改进与提高,这一技术日趋完善。2D-PAGE 的分离能力非常强大,它甚至能轻易地将细胞中的 5000 种蛋白分离开。正是由于它的强大分离能力,2D-PAGE 可用于研究基因突变、基因的表达和调控、蛋白质翻译后的修饰(如磷酸化、糖基化)。2D-PAGE 与其他生物技术,如微量蛋白质的质谱分析、自动氨基酸序列分析等技术相结合,可以快速准确地发现或鉴定蛋白质。
原理
双向聚丙烯酰胺凝胶电泳法分离蛋白质是根据蛋白质的两个一级属性-等电点和分子质量的特异性,将蛋白质混合物在电荷(等电聚焦,IEF)和分子质量(变性聚丙烯酰胺凝胺电泳,SDS-PAGE)两个水平上进行分离。2D-PAGE 的第一向电泳是等电聚焦电泳(IEF),蛋白质因所带净电荷的不同而被分离开;然后对蛋白质进行第二向电泳-SDS-PAGE,在 SDS-PAGE 中,不同分子质量的蛋白质相互间分离开。由于蛋白质的分子质量和所带净电荷是两个彼此不相关的重要性质,而 2D-PAGE 同时利用了蛋白质间在这两个性质上的差异分离蛋白质。
用途
双向聚丙烯酰胺凝胶电泳法分离蛋白质可用于研究基因突变、基因的表达和调控、蛋白质翻译后的修饰(如磷酸化、糖基化)。2D-PAGE 与其他生物技术,如微量蛋白质的质谱分析、自动氨基酸序列分析等技术相结合,可以快速准确地发现或鉴定蛋白质。
材料与仪器
步骤
双向聚丙烯酰胺凝胶电泳法分离蛋白质的基本过程可分为如下几步:
(一)准备样品
A 细胞培养、处理和收集 取对数生长期细胞(约 3 x 107~5 x 107),用胰酶消化后,清洗,1000 r/min 离心 10 min,去上清液。
B 加入 1 ml IEF 裂解缓冲液重悬细胞,反复吹打 3~5 min。4 ℃ 静置 20 min,充分裂解细胞。
C 将样品于 4 ℃、10000 g 离心 1 min,去除不溶的细胞碎片和 DNA,将上清液置于一只新的 Eppendorf 管中保存。
D 测定并调节样品蛋白浓度为 10~20 mg/ml。
E 将样品在 - 80 ℃ 条件下保存以待进一步分析。
(二)第一向等电聚焦电泳
A 配制 10 ml 凝胶溶液 将 5.4 g 的尿素(9 mol/L)、0.5 ml pH3~10 两性电解质(2%)、12 ml 30% 的丙烯酰胺/1.8% 的 N,N'-亚甲基双丙烯酰胺溶液、3.6 ml 水加入一个 50 ml 的锥形瓶(可抽真空),置锥形瓶于 37 ℃ 水浴使尿素溶解,然后抽真空排气 2 min,再加入 1.0 ml 的 20% NP-40(2%)、50 μl 10% 过硫酸铵(0.05%)、5.0 μl TEMED(0.05%),立即混匀。
B 将上述制备好的溶液倒入一个特制的平底小量筒内至所需高度。
C 将 12 支柱状胶玻璃管放入量筒内然后用封口膜将量筒密封,将量筒在室温下放置 2 h 以上,待凝胶充分聚合。
D 在准备加样前几分钟,将玻璃管从小量筒中取出,清洁管塞外的凝胶。
E 挑选 10 支玻璃管装入电泳的玻璃管支架上。
F 将上电泳槽装入真空除气过的 0.02 mol/L NaOH 溶液,使溶液液面高于玻璃管上端 0.5~1.0 cm。
G 用金属的微量注射器针头将玻璃管内的气泡除去。
H 用微量注射器将 5~10 μl 的样品蛋白加在玻璃管内的凝胶上端。
I 在下电泳槽内加满 0.01 mol/L 的磷酸溶液。
J 将玻璃管支架置入下电泳槽内,接通电源,然后调电压至 100~450 V,电泳过夜,最后将电压调至 1000 V,继续电泳 2 h。
(三)第二向 SDS-聚丙烯酰胺凝胶电泳(10%~16% 的梯度凝胶)
A 将 10 副平板胶玻璃架装人平板胶盒内。
B 按照表 8-3 顺序,配制 10% 和 16% 的凝胶溶液。
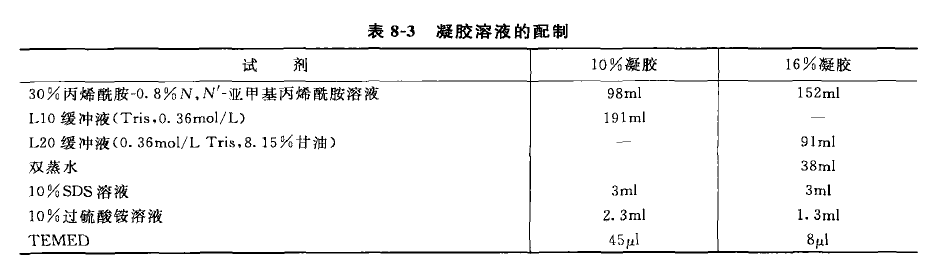 C 将梯度混合器上的 A、B 两个夹子夹紧(见图 8-6)。
C 将梯度混合器上的 A、B 两个夹子夹紧(见图 8-6)。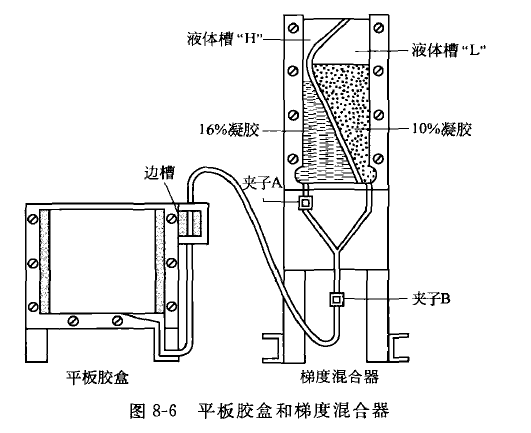
D 将 10% 和 16% 的凝胶溶液分别倒入梯度混合器的右、左两边至所需高度。
E 将夹子 B 松开,使溶液从梯度混合器的右边流入所连的管道内,等管内充满液体后,将夹子 B 夹紧,然后将夹子 A 部分放松,使液体流入连接梯度混合器的左边管道内,待管内充满液体后,将 A 夹紧。
F 先将夹子 B 完全松开,待梯度混合器两边的液面高度一致时,立即将夹子 A 松开。
G 当梯度混合器内的液体全部流入平板胶盒内时,将夹子 B 夹好。
H 在平板胶盒的边槽内,加入 50~100 ml 50% 的甘油溶液,然后将梯度混合器与平板胶盒边槽之间的管子从边槽内拔出,边槽内的甘油溶液随即流入平板胶盒内。
I 在胶面上密封覆盖一层水饱和的异丁醇或 50% 的乙醇溶液,在室温下让凝胶充分聚合至少 3 h 或放置过夜。
J IEF 结束前 1 h,将平板胶从平板胶盒内取出,清洁玻璃板外的凝胶,用去离子水将凝胶上端及表面淋洗一遍,然后将凝胶倒置,使凝胶表面和玻璃板内壁上的水引流干净。
K 加热熔化 0.5% 的琼脂糖凝胶。
L 将蛋白质分子质量标准与 0.5% 的琼脂糖凝胶混合均匀,装入一支柱状胶玻璃管内(玻璃管的一端先用封口膜封住),待胶冷却凝固数分钟。
M 等电聚焦电泳结束时,切断电源,将柱状胶玻璃管支架从下电泳槽取出,将上电泳槽内的氢氧化钠溶液倒去,用水流将柱状胶从玻璃管内冲出,装入一个 5~15 ml 的小瓶或试管内。
N 在装胶的小瓶或试管内,加入足量的平衡缓冲液,轻摇,平衡约 8 min。
O 在每块平板胶表面放 1 根柱状胶,平板胶和柱状胶之间不能有气泡。
P 将蛋白质分子质量标准琼脂糖凝胶从玻璃管内取出,切成 0.5~1.0 cm 长的小段,在每块平板胶的一端放上一段。
Q 取 0.2~0.3 ml 熔化的 0.5% 琼脂糖凝胶,将柱状胶固定在平板胶上。
R 将平板胶放入 SDS-PAGE 电泳槽内,加满电泳缓冲液。
S 接通电泳仪,以 300 mA 电流电泳过夜。
T 当溴酚蓝溶液完全电泳出平板胶时,电泳结束。平板胶可作蛋白质印迹分析,或用固定液固定后作各种蛋白质染色。
(四)凝胶固定与银染
A 将凝胶放在固定液 I 中,轻微摇荡至少 0.5 h。
B 将凝胶放在固定液 II 中,摇荡过夜或至少 0.5 h。
C 超纯水洗凝胶 3 次,每次 10 min。
D 银染液孵育 40 min。
E 显色液显色 15 min 后蛋白点显色清楚后,终止液终止显色反应。并放置于保存液中保存凝胶。
注意事项
1 整个实验过程较复杂,实验前应做好充分的准备。
2 使用高纯度试剂。
3 过硫酸铵溶液使用前配制。
4 平板胶凝胶聚合 3~12 h,不宜超过 24 h。
5 固定与显色的所有步骤均在室温进行。
来源:丁香实验