pcr扩增中出现的非特异性条带
汤姆卜丽波
如图,菌液pcr验证重组阳性克隆,目的条带大小800bp左右,先后连接到T载,和pCDNA3.1上,挑单克隆摇菌过夜后菌液pcr结果分别如图:
图1:左1为大小约800bpmarker,下图为目的基因的扩增,taq酶是TAKARA的 Extaq,引物为自己设计调试的特异性较好的,摸索好的pcr程序,取右4号做酶切连接。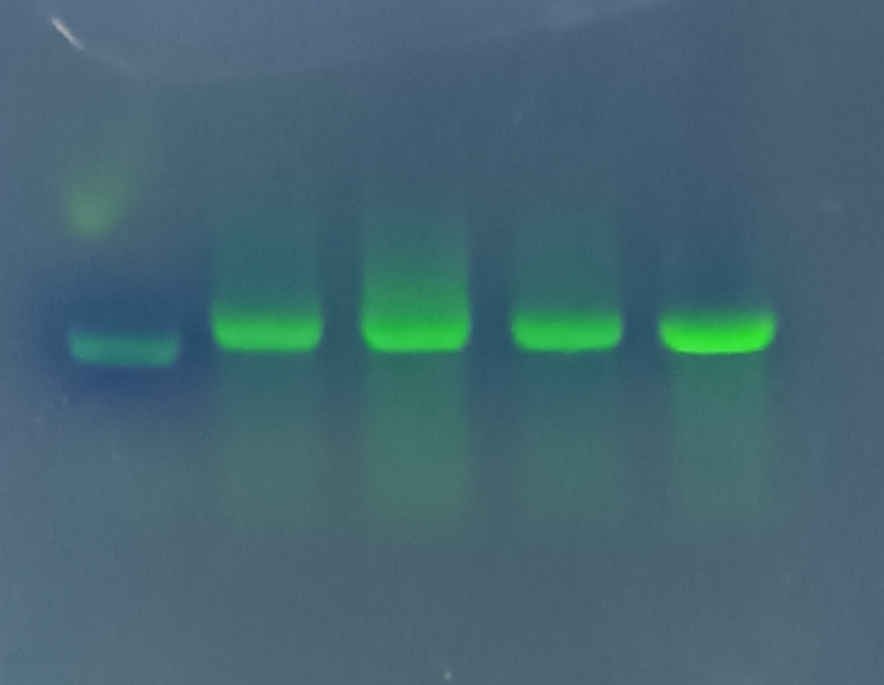
图2:图2为连接T载体后,挑单克隆菌液pcr结果,程序不变,出现了比之前做菌液pcr更明显的非特异性条带取3号连接pCDNA3.1
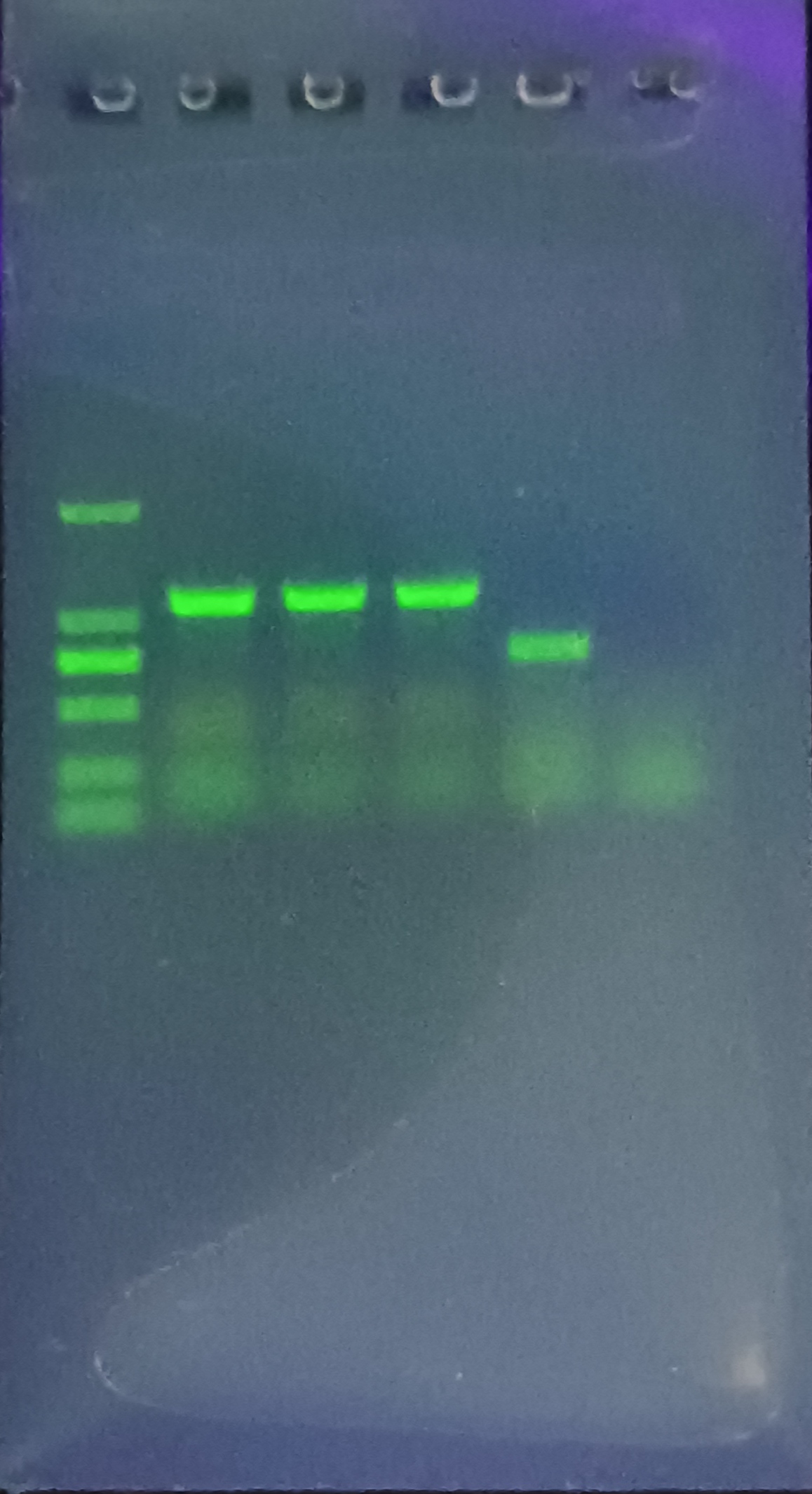
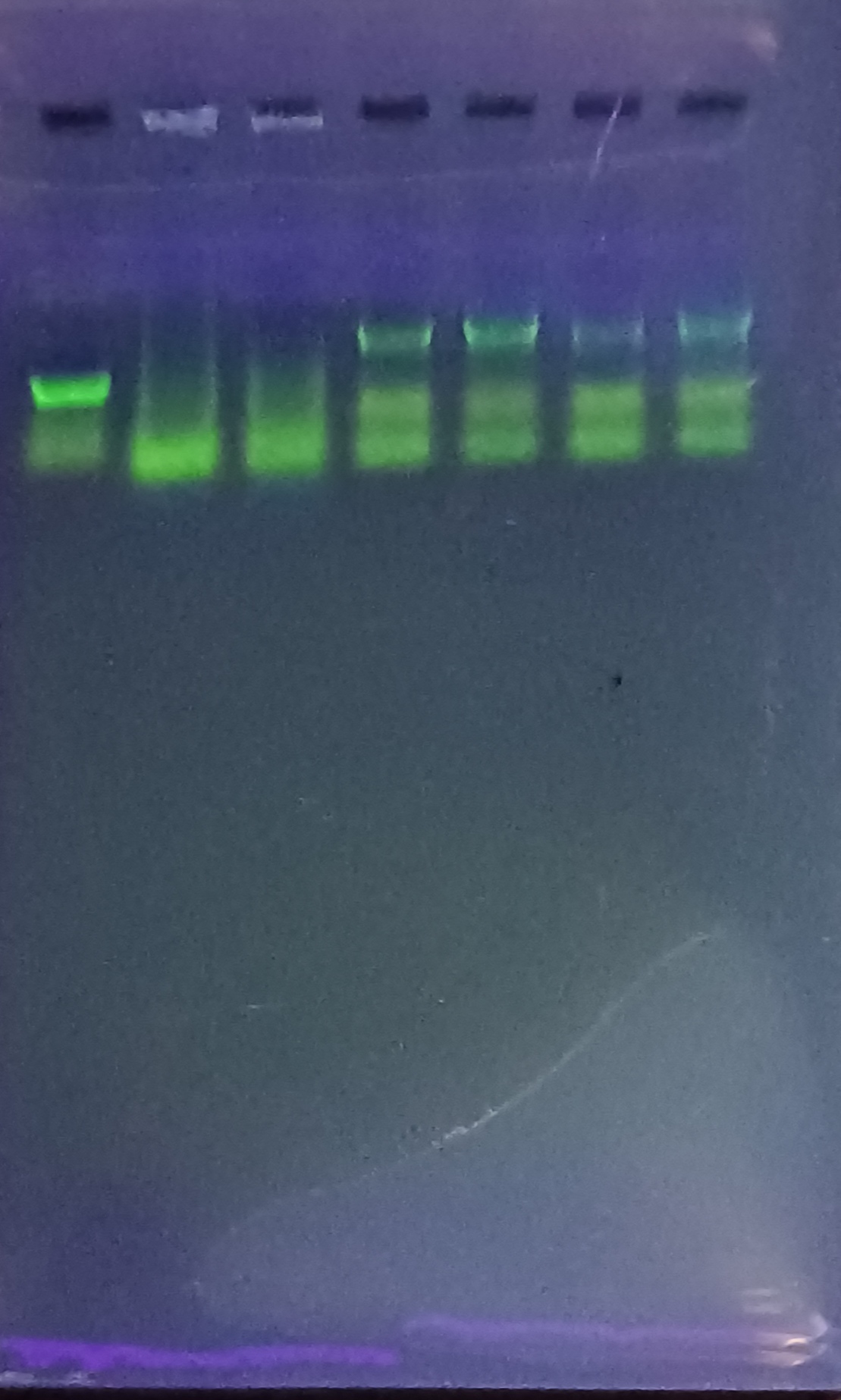 图3:3.1菌液pcr,2kmarker,程序不变,4号阳性
图3:3.1菌液pcr,2kmarker,程序不变,4号阳性
问题在于一直出现比较明显的非特异性条带,这是之前做基因重组克隆从来没有过得,程序上一直是按照以往的方法,这是怎么回事呢
4 个回答
天一湖医者
非特异性条带的出现,其原因:
一是引物与靶序列不完全互补、 或引物聚合形成二聚体.
二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数 过多有关.其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶 则不出现,酶量过多有时也会出现非特异性扩增.其对策有:
①必要时重新设计引 物.②减低酶量或调换另一来源的酶.③降低引物量,适当增加模板量,减少循环次 数.
bamboopiggy
你图一没有杂带是因为模板干净,用菌液做pcr,模板纯度比较低,是会出现杂带,只要有目的条带,就不用管杂带
Eason老歌迷
一般出现非特异性条带,我们可以用下面的办法解决,降低引物和聚合酶的用量,提高退火温度,或者是采用梯度PCR法,测试能够获得最佳结果的温度退火。如果还是不行,就试试touchdown方法,退火温度从高到低,每隔一个反应温度下降1C或者怎么样的,这样可以便于扩增出特异性更好的条带。当然,如果模板可以纯化一下,或者重新制备,也可以避免有非特异物质或者是降解的片段干扰。总之就是先优化。实在不行需要重新设计引物,并且将引物序列在NCBI里面和你的样品的基因组做一下比对,看看什么位置有可能造成非特异结合。
灵枢天问
可能是菌液的问题,菌液中含有一些非特异性组织可同步扩增
相关产品推荐




相关问答
关于丁香通
公司信息
个人用户
企业机构


