|
ABSTRACT |
|
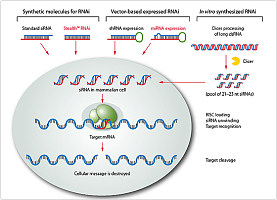
A series of protocols is presented for targeting endogenous genes in mammalian somatic cells. |
|
|
|
PROTOCOL 1: SELECTION OF SIRNA SEQUENCES |
|
To target a specific mRNA for degradation, a portion of the mRNA target sequence must be known and a segment of the target mRNA must be chosen that will be used for targeting by the cognate siRNA duplex. The siRNA selection process has recently been automated by Bingbing Yuan and Fran Lewitter at the Whitehead Institute (Cambridge, Massachusetts) and a Web Site (http://jura.wi.mit.edu/bioc/siRNA/home.php ) has been made publicly available. This software allows the user to define sequence motifs and G/C content, to search siRNAs against the human and mouse genome databases to prevent mistargeting, and to exclude single-nucleotide polymorphic sites. |
|
|
|
PROCEDURE |
-
Select the target region from the open reading frame of a desired cDNA sequence, preferably 50-100 nucleotides downstream from the start codon.
It is conceivable that 5´UTRs or 3´UTRs or regions close to the start codon are less effectively targeted by siRNAs, as these may be richer in regulatory protein-binding sites. UTR-binding proteins and/or translation initiation complexes could interfere with binding of RISC to the target RNA.
If the intent, however, is to rescue a knockdown phenotype by reintroduction of a plasmid coding for a mutant or tagged form of the targeted gene, it may be preferable to target regions in the UTRs. Preparation of rescue constructs by deletion of terminal untranslated sequences is easier than the introduction of silent mutations within the targeted region of a coding segment. In a recent survey of 3´UTR-localized targeting sites of more than 40 essential genes, we found that 3´UTRs are in fact as effectively targeted as coding regions (M. Hossbach, S. Elbashir, T. Tuschl, unpubl.).
-
Search for sequences 5´-AA(N19)UU, where N is any nucleotide, in the mRNA sequence, and ideally choose those with ~50% G/C content . Nevertheless, 32-79% G/C content has also worked well in our hands. Highly G-rich sequences should be avoided because they tend to form G-quartet structures.
If there are no 5´-AA(N19)TT motifs present in the target mRNA, search for 5´-AA(N21) or 5´-NA(N21) sequences (Figure 13.2b). Independent of the selection procedure described in Figure 13.2, synthesize the sense siRNA as 5´-(N19)TT, and the sequence of the antisense siRNA as 5´-(N´19)TT, where N´19 denotes the reverse complement sequence of N19. N19 and N´19 indicate ribonucleotides, and T indicates 2´-deoxythymidine. If the intent, however, is to also express a chemically synthesized siRNA using pol-III-based expression vectors, select the targeted sequence as 5´-NAR(N17)YNN (Figure 13.2c), where R and Y indicate purine and pyrimidine nucleotides, respectively. The sense siRNA is then synthesized accordingly as 5´-R(N17)YTT and the sequence of the antisense siRNA as 5´-R´(N´17)Y´TT.
-
Perform a BLAST search (www.ncbi.nlm.nih.gov/BLAST ) using the selected siRNA sequences as the input against EST libraries or mRNA sequences of the respective organism to ensure that only a single gene is targeted.
-
(Although optional, this step is recommended) Synthesize several siRNA duplexes to control for the specificity of the knockdown experiments.
Those siRNA duplexes that are effective for silencing should produce exactly the same phenotype. Furthermore, a nonspecific siRNA duplex may be needed as a control. It is possible to reverse the sequence of an effective siRNA duplex or to use a siRNA duplex that targets a gene absent from the selected model organism, e.g., GFP or luciferase. We have used an siRNA duplex targeting firefly luciferase as a control for targeting endogenous genes in mammalian cells because the firefly luciferase gene was not present in the targeted cells (Elbashir et al. 2002).
If the siRNA does not work, first verify that the target sequence and the cell line used are derived from the same organism. According to a recent study, there is a high probability of using the wrong cell line (Masters et al. 2001). In addition, make sure that the mRNA sequence used for selection of the siRNA duplexes is reliable; it could contain sequencing errors, mutations (e.g., in cancer cells), or polymorphisms. |
|
|
|
PROTOCOL 2: ANNEALING SIRNAS TO PRODUCE SIRNA DUPLEXES |
|
Sense and antisense siRNA strands are annealed to form a duplex prior to transfecting them into cultured cells. |
|
|
|
MATERIALS |
|
REAGENTS
-
2x Annealing buffer
-
200 mM potassium acetate
-
4 mM magnesium acetate
-
60 mM HEPES-KOH (pH 7.4)
-
Ethidium bromide solution (1% w/v) (aqueous)
-
NuSieve GTG agarose
-
Sense and antisense siRNA in H2O at a concentration >80 mM
-
Sucrose gel-loading buffer (Sambrook et al. 2001)
-
5x TBE buffer
-
450 mM Tris base
-
450 mM boric acid
-
10 mM Na2EDTA
EQUIPMENT
-
Gel electrophoresis equipment
-
UV light source
-
Water bath preset to 90ºC
|
|
PROCEDURE |
-
Prepare a 20 mM siRNA duplex solution by combining:
70 µl of 2x annealing buffer
sense siRNA to 20 mM final concentration
antisense siRNA to 20 mM final concentration
sterile H2O to a final volume of 140 µl.
-
Incubate the reaction for 1 minute at 90ºC, followed by 1 hour at 37ºC.
Store unused siRNA duplex solution frozen at -20ºC. The siRNA duplex solution can be frozen and thawed many times and does not require any further heat shock treatments. Always keep RNA solutions on ice as much as possible to reduce the rate of RNA hydrolysis.
-
To assess the completeness of the annealing reaction:
-
Separately load 1 µl of 20 mM sense and antisense siRNAs and 0.5 µl of 20 mM siRNA duplex onto a 4% NuSieve GTG agarose gel. When loading the samples, it is helpful to first dilute the samples with a few microliters of 0.5x TBE buffer and sucrose-loading buffer.
-
Run the gel in 0.5x TBE buffer at 80 V for 1 hour. NuSieve agarose is a low-melting-temperature agarose, which may melt if electro-phoresis is performed with excessive electric current.
-
Detect the RNA bands under UV light after ethidium bromide staining. Preferably, add the ethidium bromide to the 4% gel/0.5x TBE solution at a concentration of 0.4 mg/liter (4 µl of 1% ethidium bromide solution per 100 ml of gel solution) prior to casting the gel.
|
|
PROTOCOL 3: CELL CULTURE AND PREPARATION OF CELLS IN 24-WELL PLATES |
|
Transfection of cultured cells with siRNAs and downstream analysis of the knockdown cells are best performed in multiwell tissue culture plates. |
|
|
|
MATERIALS |
Products for this protocol
|
REAGENTS
-
Dulbecco's modified Eagle medium (DMEM)
-
Fetal bovine serum (FBS)
-
Mammalian cell lines (e.g., HeLa S3, HeLa SS6, COS-7, NIH-3T3, HEK 293, CHO, A431, and SKBR3)
-
Penicillin and streptomycin
-
Trypsin-EDTA solution
EQUIPMENT
-
Cell culture flask (175 ml)
-
Cell culture plate (24 well)
-
Coverslips (Optional, see Step 3.)
-
Incubator (5% CO2, humidified)
|
|
PROCEDURE |
-
Grow mammalian cell lines in a 5% CO2 humidified incubator at 37ºC in DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 µg/ml streptomycin. Passage cells regularly to maintain exponential growth.
Do not exceed a passage number of 30 after unfreezing the stock culture. The number of passages may affect DNA and siRNA transfection efficiencies. Aliquots of cells with low passage number may be stored frozen and can be thawed as needed. For general advice on cell culture, see Spector et al. (1999).
-
At least 24 hours before plasmid/siRNA transfection, trypsinize 90% confluent cells grown in a 175-ml cell culture flask with 10 ml of trypsin-EDTA.
-
Dilute the cell suspension 1:5 with fresh DMEM without antibiotics and transfer 500-ml aliquots into each well of a 24-well plate.
If immunofluorescence assays are planned, grow cells on coverslips placed at the bottom of the 24-well plates prior to addition of the cell suspension.
-
At least 24 hours after seeding the cells, ensure that a confluency of 50-80% is reached, which corresponds to 3 x 104 to 1 x 105 cells per well, depending on the cell line and its doubling time.
|
|
PROTOCOL 4: COTRANSFECTION OF LUCIFERASE REPORTER PLASMIDS WITH SIRNA DUPLEXES |
Before siRNAs are applied to knock down an endogenous gene, it may be important to establish whether the studied cells are susceptible to RNAi. It may be possible that some cell lines have lost the ability to perform RNAi or that cells derived from certain tissues do not support RNAi.
This protocol describes a reporter assay for RNAi in mammalian cells and is based on a published procedure (Elbashir et al. 2002). The quantities of reagents given below are calculated for the transfection of one well of a 24-well plate. |
|
|
|
MATERIALS |
|
REAGENTS
-
Dual-Luciferase Assay
-
LIPOFECTAMINE 2000
-
OPTI-MEM 1 medium
-
Mammalian cells
-
Plasmids
-
pGL2-Control plasmid
-
pGL3-Control plasmid
-
pRL-TK plasmid
-
siRNA duplexes (see Protocol 2)
-
∑ GL2 luciferase siRNAs sense siRNA: 5´ CGUACGCGGAAUACUUCGAdTdT
antisense siRNA: 5´ UCGAAGUAUUCCGCGUACGdTdT
-
∑ invGL2 siRNAs (inverted sequence of GL2 siRNA as nonspecific control)
sense siRNA: 5´ AGCUUCAUAAGGCGCAUGCdTdT
antisense siRNA: 5´ GCAUGCGCCUUAUGAAGCUdTdT
-
∑ GL3 luciferase siRNAs
sense siRNA: 5´ CUUACGCUGAGUACUUCGAdTdT
antisense siRNA: 5´ UCGAAGUACUCAGCGUAAGdTdT
-
∑ RL luciferase siRNAs
sense siRNA: 5´ AAACAUGCAGAAAAUGCUGdTdT
antisense siRNA: 5´ CAGCAUUUUCUGCAUGUUUdTdT
EQUIPMENT
-
Cell culture plates (24-well)
-
Incubator (37ºC, 5% CO2, humidified)
|
|
PROCEDURE |
-
On the day before transfection, culture cells in 24-well plates by completing Steps 2-4 of Protocol 3.
-
On the day of transfection, mix:
1.0 mg of pGL2-Control plasmid or 1 mg of pGL3-Control plasmid
0.1 mg of pRL-TK plasmid
0.21 mg of siRNA duplex (0.75 µl of 20 mM annealed duplex; see Protocol 2)
50 µl of OPTI-MEM 1 medium
-
In a separate tube, add 2 µl of LIPOFECTAMINE 2000 to 50 µl of OPTI-MEM 1 medium. Mix the tube gently by inverting; do not vortex. Incubate the suspension for 5 minutes at room temperature without movement.
-
Combine the solution from Step 2 with the suspension from Step 3. Mix gently by inverting the tube, and then incubate it for 20-25 minutes at room temperature to allow for formation of liposome complexes. Do not exceed a 30-minute incubation time.
-
Add the liposome complexes to the well of cells (from Step 1) without replacing the growth medium and mix gently for 15 seconds by gently rocking the plate. Incubate the plate for 20-48 hours at 37ºC in a 5% CO2 humidified incubator. If cytotoxic effects are expected from the transfection reagent, change the growth medium 5 hours after transfection.
-
To monitor luciferase activity, lyse the cells and measure luciferase expression using the Dual-Luciferase Assay according to the manufacturer's instructions.
To estimate the transfection efficiency, it is convenient to cotransfect a GFP-coding plasmid together with 0.21 mg of a siRNA duplex noncognate to GFP (e.g., invGL2) and to count the GFP-expressing cells by fluorescence microscopy. Transfection efficiencies for most cell lines described above range from 70% to 90%.
|
|
PROTOCOL 5: TRANSFECTION OF SIRNA DUPLEXES |
In the absence of reporter plasmids, siRNAs are best delivered with transfection reagents developed for delivery of antisense oligodeoxynucleotides. Such reagents are sometimes less toxic than plasmid delivery reagents and may show higher transfection efficiencies than conventional transfection reagents.
Two transfection reagents have been used predominantly in our research group: OLIGOFECTAMINE from Invitrogen and TransIT-TKO siRNA Transfection Reagent from Mirus. The quantities of reagents given below are calculated for the transfection of one well of a 24-well plate. |
|
|
|
MATERIALS |
|
REAGENTS
-
Cells (see Step 1)
-
OLIGOFECTAMINE
or
TransIT-TKO siRNA Transfection Reagent
-
OPTI-MEM 1 medium
-
siRNA duplexes (see Protocol 2)
EQUIPMENT
-
Incubator (5% CO2, humidified)
|
|
PROCEDURE |
-
The day before transfection, complete Steps 2-4 of Protocol 3, except dilute the cell suspension after trypsination of the stock culture 1:10 rather than 1:5 before transferring to the 24-well plate (see Step 3 of Protocol 3). Use a higher dilution to obtain the recommended confluency of 50% for OLIGOFECTAMINE transfection.
-
Mix 3 µl of the 20 mM siRNA duplex (0.84 mg, 60 pmoles) with 50 µl of OPTI-MEM 1.
-
In a separate tube, add 3 µl of OLIGOFECTAMINE (or 4.0 µl of TransIT-TKO) to 12 µl of OPTI-MEM 1. Mix gently and incubate it for 7-10 minutes at room temperature.
-
Slowly add the siRNA solution (Step 2) to the solution prepared in Step 3 and mix gently by inversion; do not vortex. Incubate the tube for 20-25 minutes at room temperature to allow for formation of lipid complexes; the solution will turn turbid. Then add 32 µl of fresh OPTI-MEM 1 medium to obtain a final volume of 100 µl and mix gently by inversion.
-
Add the 100 µl of lipid complexes from Step 4 to the well of cells (from Step 1) without replacing the growth medium and mix gently for 30 seconds by gently rocking the plate. Incubate the plate for 2-3 days at 37ºC in a 5% CO2 humidified incubator.
TransIT-TKO reagent is more difficult to handle than OLIGOFECTAMINE, because the concentrations required for effective transfection also cause cytotoxic effects. Typical side effects of TransIT-TKO siRNA transfection are formation of extended lamellipodia as well as oval-shaped nuclei that appear ~2 days after transfection. These effects are observed using between 4.0 and 4.5 µl of TransIT-TKO reagent. |
|
|
|
PROTOCOL 6: IMMUNOFLUORESCENCE DETECTION OF PROTEIN KNOCKDOWN |
|
The preferred way of detecting a gene knockdown is to use a specific antibody that recognizes the targeted gene product. |
|
|
|
MATERIALS |
|
REAGENTS
-
Cells from Protocol 5
-
Hoechst 33342
-
Methanol, chilled to -10ºC
-
Moviol mounting medium
-
Phosphate-buffered saline (PBS) (pH 7.1)
-
137 mM NaCl
-
7 mM Na2 HPO4
-
1.5 mM KH2 PO4
-
2.7 mM KCl
-
Specific primary and secondary antibodies
Dilute the antibodies with PBS buffer containing 0.5 mg/ml BSA (A 9706, Sigma) and 0.02% NaN3. The secondary antibody is fluorescently labeled.
EQUIPMENT
-
Ceramic rack
-
Cell culture plate (24-well) carrying knockdown cells on coverslips (from Protocol 5)
-
Coverslips
-
Filter paper
-
Incubator (37ºC)
-
Nail polish (See Step 10)
-
Petri dish (13-cm diameter) (See Step 3)
-
Slides
-
Tweezers (Dumont No. 7)
-
Upright light microscope
Alternatively, a laser-scanning microscope may be used.
|
|
PROCEDURE |
-
Fix and permeabilize the knockdown cells.
-
Use tweezers to remove the coverslips carrying the knockdown cells (from Protocol 5) from the 24-well plate.
-
Place the coverslips on a ceramic rack and then incubate them in methanol chilled to -10ºC for 6 minutes.
Methanol fixation is suitable for the detection of many cellular proteins, but the optimal fixation procedure may have to be established experimentally for each individual protein (Celis 1998; Spector et al. 1999). We recommend beginning with methanol fixation, which preserves the ultrastructure of the cell and sufficiently permeabilizes the cells for penetration of the antibody.
-
Wash the methanol-fixed coverslips three times in PBS and touch filter paper to the coverslips to remove excess PBS.
-
Place the coverslips in a wet chamber with the cells side facing up. Prepare a wet chamber by soaking filter paper in H2O and placing it into a 13-cm-diameter Petri dish. Do not allow the specimens to dry out during this procedure.
-
Add 20 µl of appropriately diluted primary antibody on top of the coverslip without touching the cells. Make sure that the solution is evenly spread out over the entire surface of the coverslip. Transfer the closed wet chamber into a 37ºC incubator and incubate for 45-60 minutes.
Antibodies are diluted with PBS containing 0.5 mg/ml bovine serum albumin and 0.02% sodium azide.
-
Place the coverslips again on the ceramic rack and wash them three times with PBS, each for 5 minutes. Touch filter paper to the coverslips to remove excess PBS, and then transfer the coverslips back into the wet chamber.
-
Add 20 µl of appropriately diluted, fluorescently labeled secondary antibody to each coverslip. Incubate the cells in the closed wet chamber for 45 minutes at 37ºC.
-
Repeat Step 5.
-
Detect the cell nuclei by chromatin staining. Add 20 µl of 1 mM Hoechst 33342 solution in PBS on top of the coverslip and incubate for 4 minutes at room temperature.
-
Repeat Step 5.
-
Mount two coverslips per slide by placing the coverslips with the cells side facing downward on a drop of Moviol mounting medium. Place a piece of filter paper on top of the slide and press gently on top of the paper to remove excess mounting medium. Glue coverslips to the slide with nail polish.
-
Examine the immunofluorescence staining and take pictures using an upright light microscope. Use identical exposure times for photographing both the silenced cells and the control-treated cells.
Alternatively, a laser-scanning microscope may be used.
|
|
|
|
PROTOCOL 7: DETECTION OF PROTEIN KNOCKDOWN BY WESTERN BLOTTING |
|
Knockdown of proteins is frequently associated with impaired cell growth or altered cell morphology, which can be monitored by phase-contrast microscopy. If no alterations in cell growth or cell morphology are observed, immunofluorescence or western blotting can be performed to analyze the depletion of the target protein. |
|
|
|
MATERIALS |
Products for this protocol
|
REAGENTS
-
Blocking solution (5% milk powder in TBST [pH 7.4])
-
Dulbecco's modified Eagle medium (DMEM)
-
ECL (enhanced chemiluminescent) detection kit
-
Electrotransfer buffer
-
25 mM Tris
-
192 mM glycine
-
0.01% SDS
-
20% methanol
-
2x Laemmli SDS sample buffer
-
Phosphate-buffered saline (PBS) (pH 7.1)
-
137 mM NaCl
-
7 mM Na2 HPO4
-
1.5 mM KH2 PO4
-
2.7 mM KCl
-
Ponceau S stain (Sigma)
-
Primary antibody - See Step 8. If necessary, dilute the antibody in TBST
-
Secondary antibody
-
Horseradish peroxidase (HRP)-conjugated rabbit anti-mouse or HRP-conjugated swine
-
anti-rabbit antibodies
-
siRNA-treated cells cultivated in 24-well plates (from Protocol 5)
-
TBST (pH 7.4)
-
0.2% Tween-20
-
20 mM Tris-HCl
-
150 mM NaCl
-
Trypsin-EDTA solution
EQUIPMENT
-
Centrifugation tube (1.5 ml)
-
Centrifuge
-
Electrotransfer equipment
-
Enhanced chemiluminescent detection equipment
-
Nitrocellulose membrane
-
SDS-polyacrylamide gel electrophoresis equipment
-
Water bath (boiling)
|
|
PROCEDURE |
-
Remove the tissue culture medium from the siRNA-treated cells cultivated in 24-well plates (from Protocol 5). Rinse the cells once with 200 µl of PBS, and add 200 µl of trypsin-EDTA. Incubate for 1 minute at 37ºC; suspend the cells and add 800 µl of DMEM medium to quench the trypsin.
-
Transfer the suspended cells to a chilled 1.5-ml centrifugation tube. Collect the cells by centrifugation at 3000 rpm (700g) for 4 minutes at 4ºC. Resuspend the cell pellet in ice-cold PBS and centrifuge again.
-
Remove the supernatant and add 25 µl of 90ºC 2x concentrated Laemmli SDS sample buffer to the cell pellet obtained from one well of a 24-well plate. Incubate the sample for 3 minutes in a boiling water bath and vortex.
-
Separate the proteins by SDS-polyacrylamide gel electrophoresis using an acrylamide concentration appropriate to resolve the molecular weight of the targeted protein (Sambrook et al. 2001).
-
We have separated proteins on minigels, which were run at a constant 10 mA
-
Transfer proteins from the gel to a nitrocellulose membrane using electrotransfer buffer. Our minigels are electroblotted onto the membrane using a Bio-Rad Trans-Blot cell at 333 mA for 30 minutes in the cold room.
-
Verify the protein transfer by Ponceau S staining of the transfer membrane.
-
Incubate the membrane in blocking solution for 1 hour at 37ºC.
-
Replenish the blocking solution with fresh blocking solution and add the primary antibody at the appropriate dilution. Incubate for 1-2 hours at 37ºC.
-
Wash the blot four times with TBST for 10 minutes.
-
For ECL detection, incubate the blot with either HRP-conjugated rabbit anti-mouse or HRP-conjugated swine anti-rabbit antibodies at a dilution of 1:20,000 in blocking solution for 1-2 hours at 37ºC.
-
Perform ECL detection according to the protocol described by the manufacturer.
|
|
|
|
REFERENCES |
Celis J.E. 1998. Cell biology: A Laboratory Handbook , Vol. 2. Academic Press, San Diego.
Elbashir S.M., Harborth j., Lendeckel W., and Tuschl T. 2002. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 26: 199-213.
Masters J.R., Thomson J.A., Daly-Burns B., Reid Y.A., Dirks W.G., Packer P., Toji L.H., Ohno T., Tanabe H., Arlett C.F., Kelland L.R., Harrison M., Virmani A., Ward T.H., Ayres K.L., and Debenham P.G. 2001. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl. Acad. Sci . 98: 8012-8017.
Sambrook J., Fritsch E., and Maniatis T. 2001. Molecular Cloning: A Laboratory Manual , Second Edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Spector D.L., Goldman R.D., and Leinwand L.A. 1999. Cells: A Laboratory Manual , Vols. I-III. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. |