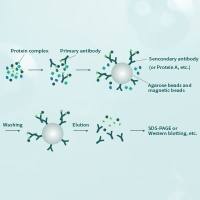
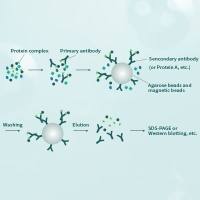
Co-immunoprecipitation assays
互联网
co-IP assays can be performed between endogenous proteins or transiently or stably expressed exogenous - usually tagged - proteins. The advantage to using endogenous proteins is the avoidance of protein overexpression and tagging that can lead to artifacts. The advantage to using exogenously expressed tagged proteins is the generally high specificity of the anti-tag antibody and the ease with which a negative control can be performed in which the exogenous protein is lacking.
Getting cells ready for IP:
Transient transfection (optional):
Three days before the IP:
Trypsinize a confluent 10-cm plate of cells and plate 0.25 ml cells plus 1.75 ml medium per 3.5-cm plate (for multiple plates, mix cells after trypsinizing and plate from the mixture to get more homogenous plates). This can be scaled up or down as needed.
Two days before the IP:
When plates are 30-50% confluent transfect cells with plasmids expressing exogneous tagged proteins using a transfection reagent as described by the manufacturer. Incubate cells 36-48 hours.
Immunoprecipitation:
Coupling the antibody to beads:
One day before IP:
Take protein A Sepharose (for rabbit and human polyclonal and most mouse monoclonal antibodies) or protein G sepharose (for mouse polyclonal or IgG1 monoclonal Abs) up in NET-2 at 100 mg/ml.
Swell beads by nutating 30 mins at RT. Wash 2x with NET-2 and resuspend at 100 mg/ml.
For each IP reaction, mix 40 μl of 100 mg/ml protein A Sepharose with 2-20 μl serum (should be titrated) in 400 μl NET-2/0.5 mM PMSF/2 μg/ml aprotinin/2 μg/ml leupeptin.
.
Nutate over night at 4?C.
Alternatively, pre-coupled antibodies can be used directly.
If the antibody used for Western blots is of the same animal origin as the antibody used for IP, the IP antibody may have to be cross-linked to the beads (see Harlow and Lane, "Using antibodies - A laboratory manual).
Immunoprecipitation:
Getting the coupled antibody ready:
Wash Protein A Sepharose with conjugated antibodies or beads with pre-coupled antibodies three times with NET-2.
For each wash, spin 2,000 rpm, 1 min and remove supernatant.
Preparation of cell extract:
Wash cells carefully with 2 ml PBS.
Add 1 ml PBS and scrape off cells using a rubber policeman for strongly attached cells or by pipetting up and down for loosely attached cells.
Transfer to eppendorf tube.
Spin 1,000 rpm, 10 min (not faster!).
Remove PBS.
Prepare a cell lysate by resuspending cells in ice-cold gentle lysis buffer (use approximatley 400 μl per 2x106 human cells, corresponding to one 3.5-cm well; the lysis buffer used should be optimized for the specific application).
Incubate on ice, 5 min.
Add NaCl to 150 mM. (Optional: Add 5 μl 10 mg/ml RNase A). Incubate on ice 10 min.
Spin 14,000 rpm, 4?C, 15 min.
Transfer 50 μl supernatant to 50 μl of SDS LB (Total extract control). Store at -20C.
Immunoprecipitation:
Transfer remainder of supernatant to tube containing washed antibody conjugate.
Nutate at 4?C, ≥2-4h.
Wash beads 8 times with 1 ml NET-2 (ice-cold). Spin 1,000 rpm, 1 min for each wash.
After last wash, remove all wash buffer with a pipette.
Elution:
Elute by adding SDS LB directly to beads, or by gently shaking beads at 4?C for 30 mins in NET-2 containing 0.2 mg/ml of a competing peptide (e.g. FLAG or myc peptides - serial elutions increase the yield).
Western blot:
Run SDS gel.
Cut four pieces of 3MM papers and a piece of nitrocellulose membrane at 8x11 cm (or 15x20cm for large gel).
Prepare 1 liter (or 4 liters for large gel) of transfer buffer: 20% methanol, 3.1 g/l Trizma base, 14.4 g/l Glycine. Pre-cool in cold room.
Transfer gel to a piece of 3MM and place on top of a piece of 3MM under transfer buffer. Place membrane on top of gel followed by two pieces of 3MM. Remove bubbles by rolling a pipette over the "sandwich".
Transfer to transfer box and fill with transfer buffer.
Transfer at 80V 3-4 hours or 20V over night in cold room. For very small proteins (<20kD) transfer for 1 hour only as they can migrate through the membrane.
(Optional: Incubate membrane with Ponceau Red to visualize transferred protein. Wash with Blot buffer)
Transfer membrane to Blot buffer/10% milk.
Incubate at RT ≥ 1 hour or at 4C over night.
Incubate at RT 1 h at RT to several days at 4?C with primary antibody in Blot buffer/5% milk.
Wash 2x5 min with Blot buffer.
Incubate at RT 1-4 h with secondary antibody in Blot buffer/5%milk. (Use Pierce HRP-coupled secondary Abs at 1:20,000)
Wash at least 3x10 min with blot buffer.
Drip off, but don''t dry out.
Incubate with chemiluminescence reagent 5 min.
Drip off, but don''t dry out!!
Wrap in Saram Wrap. Use autorad tape (or other means to orient film)
Expose to film 1 min.
Develop, and evaluate for longer or shorter exposure.
(The signal has a half-life of 5-10 mins.)
Stripping the membrane:
Incubate in water 15 min.
Incubate in 0.1M NaOH, 15 min.
Incubate in water 15 min.
Block in Blot buffer/10% milk, 4C, o/n.
Buffers:
Hypotonic gentle lysis buffer:
10 mM Tris-HCl pH 7.5
10 mM NaCl (use higher salt for interactions that are disrupted at low salt)
2 mM EDTA (omit for interactions that require divalent metal ions)
0.1% Triton-X100 (can be exchanged by other detergents, such as the milder Igepal/NP40)
1 mM PMSF
2 μg/ml aprotinin
2 μg/ml leupeptin
0.1 units/μl RNasin
NET-2:
50 mM Tris-HCl pH7.5
150 mM NaCl
0.05% Triton-X100
SDS LB:
see "Sambrook et al."
Blot buffer:
100 mM Tris HCl pH 7.5
150 mM NaCl
0.1% Tween 20